
On the Efficiency of the Density Functional Theory (DFT)-Based Computational Protocol for 1H and 13C Nuclear Magnetic Resonance (NMR) Chemical Shifts of Natural Products: Studying the Accuracy of the pecS-n (n = 1, 2) Basis Sets


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Brief Notes on PEC-Generated Contraction Coefficients
2.2. Benchmark Calculations
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Semenov, V.A.; Krivdin, L.B. Computational NMR of natural products. Russ. Chem. Rev. 2022, 91, RCR5027. [Google Scholar] [CrossRef]
- Hehre, W.; Klunzinger, P.; Deppmeier, B.; Driessen, A.; Uchida, N.; Hashimoto, M.; Fukushi, E.; Takata, Y. Efficient Protocol for Accurately Calculating 13C Chemical Shifts of Conformationally Flexible Natural Products: Scope, Assessment, and Limitations. J. Nat. Prod. 2019, 82, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Gale, J.D. SIESTA: A Linear-Scaling Method for Density Functional Calculations. In Computational Methods for Large Systems: Electronic Structure Approaches for Biotechnology and Nanotechnology; Reimers, J.R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; Chapter 2; pp. 45–75. [Google Scholar]
- Keal, T.W.; Tozer, D.J.; Helgaker, T. GIAO shielding constants and indirect spin–spin coupling constants: Performance of density functional methods. Chem. Phys. Lett. 2004, 391, 374–379. [Google Scholar] [CrossRef]
- Laskowski, R.; Blaha, P.; Tran, R. Assessment of DFT functionals with NMR chemical shifts. Phys. Rev. 2013, 87, 195130. [Google Scholar] [CrossRef]
- Zahn, S.L.V.; Hammerich, O.; Hansen, P.E.; Sauer, S.P.A. The best density functional theory functional for the prediction of 1H and 13C chemical shifts of protonated alkylpyrroles. J. Comput. Chem. 2021, 42, 1248–1262. [Google Scholar] [CrossRef]
- Vila, J.A.; Baldoni, H.A.; Scheraga, H.A. Performance of Density Functional Models to Reproduce Observed 13C Chemical Shifts of Proteins in Solution. J. Comput. Chem. 2009, 30, 884–892. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, A.; Xu, X.; Yan, Y. OPBE: A promising density functional for the calculation of nuclear shielding constants. Chem. Phys. Lett. 2006, 421, 383–388. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward chemical accuracy in the computation of NMR shieldings: The PBE0 model. Chem. Phys. Lett. 1998, 298, 113–119. [Google Scholar] [CrossRef]
- Perdew, J.P.; Schmidt, K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1–20. [Google Scholar] [CrossRef]
- Helgaker, T.; Jaszuński, M.; Ruud, K. Ab initio methods for the calculation of NMR shielding and indirect spin–spin coupling constants. Chem. Rev. 1999, 99, 293–352. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kutzelnigg, W. Theory of Magnetic Susceptibilities and NMR Chemical Shifts in Terms of Localized Quantities. Isr. J. Chem. 1980, 19, 193–200. [Google Scholar] [CrossRef]
- Schindler, M.; Kutzelnigg, W. Theory of magnetic susceptibilities and NMR chemical shifts in terms of localized quantities. II. Application to some simple molecules. J. Chem. Phys. 1982, 76, 1919–1933. [Google Scholar] [CrossRef]
- Jensen, F. Basis Set Convergence of Nuclear Magnetic Shielding Constants Calculated by Density Functional Methods. J. Chem. Theory Comput. 2008, 4, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Polarization consistent basis sets: Principles. J. Chem. Phys. 2001, 115, 9113–9125. [Google Scholar] [CrossRef]
- Jensen, F. Polarization consistent basis sets. II. Estimating the Kohn–Sham basis set limit. J. Chem. Phys. 2002, 116, 7372–7379. [Google Scholar] [CrossRef]
- Jensen, F. Polarization consistent basis sets. III. The importance of diffuse functions. J. Chem. Phys. 2002, 117, 9234–9240. [Google Scholar] [CrossRef]
- Jensen, F.; Helgaker, T. Polarization consistent basis sets. V. The elements Si–Cl. J. Chem. Phys. 2004, 121, 3463–3470. [Google Scholar] [CrossRef]
- Jensen, F. Segmented Contracted Basis Sets Optimized for Nuclear Magnetic Shielding. J. Chem. Theory Comput. 2015, 11, 132–138. [Google Scholar] [CrossRef]
- Jensen, F. Unifying General and Segmented Contracted Basis Sets. Segmented Polarization Consistent Basis Sets. J. Chem. Theory Comput. 2014, 10, 1074–1085. [Google Scholar] [CrossRef]
- Rusakov, Y.; Rusakova, I.L. New pecS-n (n = 1, 2) basis sets for quantum chemical calculations of the NMR chemical shifts of H, C, N, and O nuclei. J. Chem. Phys. 2022, 156, 244112. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.; Rusakova, I.L. New efficient pecS-n (n = 1, 2) basis sets for quantum chemical calculations of 31P NMR chemical shifts. Phys. Chem. Chem. Phys. 2023, 25, 18728–18741. [Google Scholar] [CrossRef]
- Rusakov, Y.Y.; Rusakova, I.L. An efficient method for generating propertyenergy consistent basis sets. New pecJ-n (n = 1, 2) basis sets for high-quality calculations of indirect nuclear spin–spin coupling constants involving 1H, 13C, 15N, and 19F nuclei. Phys. Chem. Chem. Phys. 2021, 23, 14925–14939. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.; Rusakova, I.L. New pecJ-n (n = 1, 2) Basis Sets for Selenium Atom Purposed for the Calculations of NMR Spin–Spin Coupling Constants Involving Selenium. Int. J. Mol. Sci. 2023, 24, 7841. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.; Rusakova, I.L. New pecJ-n (n = 1, 2) Basis Sets for High-Quality Calculations of Indirect Nuclear Spin–Spin Coupling Constants Involving 31P and 29Si: The Advanced PEC Method. Molecules 2022, 27, 6145. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Verbeke, J.; Cools, R. The Newton-Raphson method. Int. J. Math. Educ. Sci. Technol. 1995, 26, 177–193. [Google Scholar] [CrossRef]
- Helgaker, T.; Wilson, P.J.; Amos, R.D.; Handy, N.C. Nuclear shielding constants by density functional theory with gauge including atomic orbitals. J. Chem. Phys. 2000, 113, 2983–2989. [Google Scholar] [CrossRef]
- Semenov, V.A.; Krivdin, L.B. DFT computational schemes for 1H and 13C NMR chemical shifts of natural products, exemplified by strychnine. Magn. Reson. Chem. 2020, 58, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Frédérich, M.; Tits, M.; Hayette, M.-P.; Brandt, V.; Penelle, J.; DeMol, P.; Llabrès, G.; Angenot, L. 10’-Hydroxyusambarensine, a New Antimalarial Bisindole Alkaloid from the Roots of Strychnos usambarensis. J. Nat. Prod. 1999, 62, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Dassonneville, L.; Wattez, N.; Mahieu, C.; Colson, P.; Houssier, C.; Frédérich, M.; Tits, M.; Angenot, L.; Bailly, C. The plant alkaloid usambarensine intercalates into DNA and induces apoptosis in human HL60 leukemia cells. Anticancer Res. 1999, 19, 5245–5250. [Google Scholar] [PubMed]
- Kobayashi, M.; Aoki, S.; Gato, K.; Matsunami, K.; Kurosu, M.; Kitagawa, I. Marine Natural Products. XXXIV. Trisindoline, a New Antibiotic Indole Trimer, Produced by a Bacterium of Vibrio sp. Separated from the Marine Sponge Hyrtios altum. Chem. Pharm. Bull. 1994, 42, 2449–2451. [Google Scholar] [CrossRef] [PubMed]
- Capdevielle, P.; Maumy, M. 3-Oxo 3H-indole from dioxygen copper-catalyzed oxidation of indole: One-flask synthesis of 2-dialkylamino 3-oxo 3H-indoles. Tetrahedron Lett. 1993, 34, 2953–2956. [Google Scholar] [CrossRef]
- Takeshige, Y.; Egami, Y.; Wakimoto, T.; Abe, I. Production of indole antibiotics induced by exogenous gene derived from sponge metagenomes. Mol. BioSyst. 2015, 11, 1290–1294. [Google Scholar] [CrossRef]
- El-Desoky, A.H.; Kato, H.; Eguchi, K.; Kawabata, T.; Fujiwara, Y.; Losung, F.; Mangindaan, R.E.P.; de Voogd, N.J.; Takeya, M.; Yokosawa, H.; et al. Acantholactam and Pre-neo-kauluamine, Manzamine-Related Alkaloids from the Indonesian Marine Sponge Acanthostrongylophora ingens. J. Nat. Prod. 2014, 77, 1536–1540. [Google Scholar] [CrossRef]
- Hirasawa, Y.; Miyama, S.; Takahiro, H.; Koyama, K.; Rahman, A.; Kusumawati, I.; Zaini, N.C.; Morita, H. Alasmontamine A, A First Tetrakis Monoterpene Indole Alkaloid from Tabernaemontana elegans. Org. Lett. 2009, 11, 5718–5721. [Google Scholar] [CrossRef]
- Aberham, A.; Cicek, S.S.; Schneider, P.; Stuppner, H. Analysis of Sesquiterpene Lactones, Lignans, and Flavonoids in Wormwood (Artemisia absinthium L.) Using High-Performance Liquid Chromatography (HPLC)-Mass Spectrometry, Reversed Phase HPLC, and HPLC-Solid Phase Extraction-Nuclear Magnetic Resonance. J. Agric. Food Chem. 2010, 58, 10817–10823. [Google Scholar] [CrossRef]
- Chang, R.; Lotti, V.; Monaghan, R.; Birnbaum, J.; Stapley, E.; Goetz, M.; Albers-Schonberg, G.; Patchett, A.; Liesch, J.; Hensens, O.; et al. A Potent Nonpeptide Cholecystokinin Antagonist Selective for Peripheral Tissues Isolated from Aspergillus alliaceus. Science 1985, 230, 177–179. [Google Scholar] [CrossRef]
- Sun, H.H.; Byard, S.J.; Cooper, R. Revised NMR assignments for the cholecystokinin antagonist asperlicin. J. Antibiot. 1994, 47, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Geiger, W.B.; Conn, J.E.; Waksman, S.A. Chaetomin, a New Antibiotic Substance Produced by Chaetomium cochliodes: II. Isolation and Concentration. J. Bacteriol. 1944, 48, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Taylor, A. Sporidesmins. Part XIII. Ovine III-thrift in Nova Scotia. Part III. The characterisation of chetomin a toxic metabolite of Chaetomium cochliodes and Chaetomium globosum. J. Chem. Soc. Perkin Trans. 1972, 1, 472–479. [Google Scholar] [CrossRef]
- Brewer, D.; Mclnnes, A.G.; Smith, D.G.; Taylor, A.; Walter, J.A.; Loosli, H.R.; Kis, L. Sporidesmins. Part 16. The Structure of Chetomin, a Toxic Metabolite of Chaetomium cochliodes, by Nitrogen-15 and Carbon-13 Nuclear Magnetic Resonance Spectroscopy. J. Chem. Soc. Perkin Trans. 1978, 1, 1248–1251. [Google Scholar] [CrossRef]
- Xu, M.; Dong, P.; Tian, X.; Wang, C.; Huo, X.; Zhang, B.; Wu, L.; Deng, S.; Ma, X. Drug interaction study of natural steroids from herbs specifically toward human UDP-glucuronosyltransferase (UGT) 1A4 and their quantitative structure activity relationship (QSAR) analysis for prediction. Pharmacol. Res. 2016, 110, 139–150. [Google Scholar] [CrossRef]
- Liu, M.-J.; Wang, Z.; Ju, Y.; Zhou, J.-B.; Wang, Y.; Wong, R.N.-S. The mitotic-arresting and apoptosis-inducing effects of diosgenyl saponins on human leukemia cell lines. Biol. Pharm. Bull. 2004, 27, 1059–1065. [Google Scholar] [CrossRef]
- Chen, X.-B.; Wang, Z.-L.; Yang, Q.-Y.; Zhao, F.-Y.; Qin, X.-L.; Tang, X.-E.; Du, J.-L.; Chen, Z.-H.; Zhang, K.; Huang, F.-J. Diosgenin glucoside protects against spinal cord injury by regulating autophagy and alleviating apoptosis. Int. J. Mol. Sci. 2018, 19, 2274. [Google Scholar] [CrossRef]
- Wu, Y.; Ye, F.; Lu, Y.; Yong, H.; Yin, R.; Chen, B.; Yong, Y. Diosgenin glucoside protects against myocardial injury in diabetic mice by inhibiting RIP140 signaling. Am. J. Transl. Res. 2018, 10, 3742–3749. [Google Scholar]
- Chen, T.; Jiang, W.; Zhang, H.; You, X.; Liu, M.; Wang, L.; Xiang, P.; Xu, L.; Zheng, D.; Zhang, X.; et al. Protective effect of trillin against ethanol-induced acute gastric lesions in an animal model. RSC Adv. 2016, 6, 20081–20088. [Google Scholar] [CrossRef]
- Feng, B.; Kang, L.-P.; Ma, B.-P.; Quan, B.; Zhou, W.-B.; Wang, Y.-Z.; Zhao, Y.; Liu, Y.-X.; Wang, S.-Q. The substrate specificity of a glucoamylase with steroidal saponin-rhamnosidase activity from Curvularia lunata. Tetrahedron 2007, 63, 6796–6812. [Google Scholar] [CrossRef]
- Chai, X.-Y.; Xu, Z.-R.; Bai, C.-C.; Zhou, F.-R.; Tu, P.-F. A new seco-friedelolactone acid from the bark and twigs of Itoa orientalis. Fitoterapia 2009, 80, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Ebada, S.S.; Wray, V.; de Voogd, N.J.; Deng, Z.; Lin, W.; Proksch, P. Two New Jaspamide Derivatives from the Marine Sponge Jaspis splendens. Mar. Drugs 2009, 7, 435–444. [Google Scholar] [CrossRef]
- Hallock, Y.F.; Cardellina II, J.H.; Schtifer, M.; Bringmann, G.; Franqois, G.; Boyd, M.R. Korundamine A, a novel hiv-inhibitory and antimalarial “hybrid” naphthylisoquinoline alkaloid heterodimer from ancistrocladus korupensis. Bioorg. Med. Chem. Lett. 1998, 8, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Zhang, C.; Luo, T.; Wang, J.; Tang, Y.; Chen, Z.; Yu, L. Limonin: A Review of Its Pharmacology, Toxicity, and Pharmacokinetics. Molecules 2019, 24, 3679. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.D.; Kwon, H.C.; Yang, M.C.; Lee, K.H.; Choi, S.U.; Lee, K.R. Isolation of Limonoids and Alkaloids from Phellodendron amurense and Their Multidrug Resistance (MDR) Reversal Activity. Arch. Pharm. Res. 2007, 30, 58–63. [Google Scholar] [CrossRef]
- Frédérich, M.; Jacquier, M.-J.; Thépenier, P.; De Mol, P.; Tits, M.; Philippe, G.; Delaude, C.; Angenot, L.; Zèches-Hanrot, M. Antiplasmodial Activity of Alkaloids from Various Strychnos Species. J. Nat. Prod. 2002, 65, 1381–1386. [Google Scholar] [CrossRef]
- Massiot, G.; Massoussa, B.; Thepenier, P.; Jaquier, M.-J.; le Men-Olivier, L.; Delaude, C. Structure of matopensine, a novel dimeric indole alkaloid from Strychnos species. Heterocycles 1983, 20, 2339–2342. [Google Scholar] [CrossRef]
- Massiot, G.; Massoussa, B.; Jacquier, M.-J.; Thépénier, P.; Le Men-Olivier, L.; Delaude, C.; Verpoorte, R. Alkaloids from roots of strychnos matopensis. Phytochemistry 1988, 27, 3293–3304. [Google Scholar] [CrossRef]
- Fukai, T.; Hano, Y.; Hirakura, K.; Nomura, T.; Uzawa, J.; Fukushima, K. Structures of two natural hypotensive Diels-Alder type adducts, mulberrofurans F and G, from the cultivated mulberry tree (Morus lhou KOIDZ.). Chem. Pharm. Bull. 1985, 33, 3195–3204. [Google Scholar] [CrossRef]
- Mongkolvisut, W.; Sutthivaiyakit, S. Antimalarial and Antituberculous Poly-O-acylated Jatrophane Diterpenoids from Pedilanthus tithymaloides. J. Nat. Prod. 2007, 70, 1434–1438. [Google Scholar] [CrossRef]
- Okuda, T.; Yoshida, T.; Hatano, T. New Methods of Analyzing Tannins. J. Nat. Prod. 1989, 52, 1–31. [Google Scholar] [CrossRef]
- Feldman, K.S.; Ensel, S.M. Ellagitannin Chemistry. Preparative and Mechanistic Studies of the Biomimetic Oxidative Coupling of Galloyl Esters. J. Am. Chem. Soc. 1994, 116, 3357–3366. [Google Scholar] [CrossRef]
- Feldman, K.S.; Smith, R.S. Ellagitannin Chemistry. First Total Synthesis of the 2,3- and 4,6-Coupled Ellagitannin Pedunculagin. J. Org. Chem. 1996, 61, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.-F.; Xu, M.; Yang, C.-R.; Xu, M.; Zhang, Y.-J. Phenolic Antioxidants from the Leaves of Camellia pachyandra Hu. J. Agric. Food Chem. 2010, 58, 8820–8824. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Wang, X.-Y.; Ren, Z.; Qian, C.-W.; Li, Y.-C.; Kaio, K.; Wang, Q.-D.; Zhang, Y.; Zheng, L.-Y.; Jiang, J.-H.; et al. Phyllaemblicin B inhibits Coxsackie virus B3 induced apoptosis and myocarditis. Antivir. Res. 2009, 84, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Tanaka, T.; Iwamoto, Y.; Yang, C.-R.; Kouno, I. Novel Norsesquiterpenoids from the Roots of Phyllanthus emblica. J. Nat. Prod. 2000, 63, 1507–1510. [Google Scholar] [CrossRef]
- Januário, A.H.; Filho, E.R.; Pietro, R.C.L.R.; Kashima, S.; Sato, D.N.; França, S.C. Antimycobacterial physalins from Physalis angulata L. (Solanaceae). Phytother. Res. 2002, 16, 445–448. [Google Scholar] [CrossRef]
- Chen, H.; Wang, W.; Yu, S.; Wang, H.; Tian, Z.; Zhu, S. Procyanidins and Their Therapeutic Potential against Oral Diseases. Molecules 2022, 27, 2932. [Google Scholar] [CrossRef]
- Khan, M.L.; Haslam, E.; Williamson, M.P. Structure and conformation of the procyanidin B-2 dimer. Magn. Reson. Chem. 1997, 35, 854–858. [Google Scholar] [CrossRef]
- Teitelbaum, D.T.; Ott, J.E. Acute Strychnine Intoxication. Clin. Toxicol. 1970, 3, 267–273. [Google Scholar] [CrossRef]
- Martin, G.E.; Hadden, C.E.; Crouch, R.C.; Krishnamurthy, V.V. ACCORD-HMBC: Advantages and disadvantages of static versus accordion excitation. Magn. Reson. Chem. 1999, 37, 517–528. [Google Scholar] [CrossRef]
- Tchinda, A.T.; Jansen, O.; Nyemb, J.-N.; Tits, M.; Dive, G.; Angenot, L.; Frederich, M. Strychnobaillonine, an Unsymmetrical Bisindole Alkaloid with an Unprecedented Skeleton from Strychnos icaja Roots. J. Nat. Prod. 2014, 77, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wijeratne, E.M.K.; Bashyal, B.P.; Zhan, J.; Seliga, C.J.; Liu, M.X.; Pierson, E.E.; Pierson, L.S.; VanEtten, H.D.; Gunatilaka, A.A.L. Cytotoxic and Other Metabolites of Aspergillus Inhabiting the Rhizosphere of Sonoran Desert Plants. J. Nat. Prod. 2004, 67, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, G.E.; Randall, E.W.; Hull, W.E.; Gattegno, D.; Conti, F. Qualitative aspects of hydrogen-deuterium exchange in the proton, carbon-13, and nitrogen-15 nuclear magnetic resonance spectra of viomycin in aqueous solution. Biochemistry 1978, 17, 3986–3992. [Google Scholar] [CrossRef]
- Cimino, P.; Gomez-Paloma, L.; Duca, D.; Riccio, R.; Bifulco, G. Comparison of different theory models and basis sets in the calculation of 13C NMR chemical shifts of natural products. Magn. Reson. Chem. 2004, 42, S26–S33. [Google Scholar] [CrossRef]
- Casella, G.; Bagno, A.; Komorovsky, S.; Repisky, M.; Saielli, G. Four-Component Relativistic DFT Calculations of 13C Chemical Shifts of Halogenated Natural Substances. Chem. Eur. J. 2015, 21, 18834–18840. [Google Scholar] [CrossRef]
- Semenov, V.A.; Samultsev, D.O.; Krivdin, L.B. 1H and 13C NMR spectra of Strychnos alkaloids: Selected NMR updates. Int. J. Quantum Chem. 2020, 120, e26348. [Google Scholar] [CrossRef]
- Aidas, K.; Angeli, C.; Bak, K.L.; Bakken, V.; Bast, R.; Boman, L.; Christiansen, O.; Cimiraglia, R.; Coriani, S.; Dahle, P.; et al. The Dalton quantum chemistry program system. WIREs Comput. Mol. Sci. 2014, 4, 269–284. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-1: Maestro; Schrödinger, LLC.: New York, NY, USA, 2018; Available online: https://www.schrodinger.com/freemaestro (accessed on 8 August 2023).
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cances, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Irkutsk Supercomputer Center of SB RAS. Irkutsk: ISDCT SB RAS. Available online: https://hpc.icc.ru (accessed on 27 July 2023).






{kind=link}
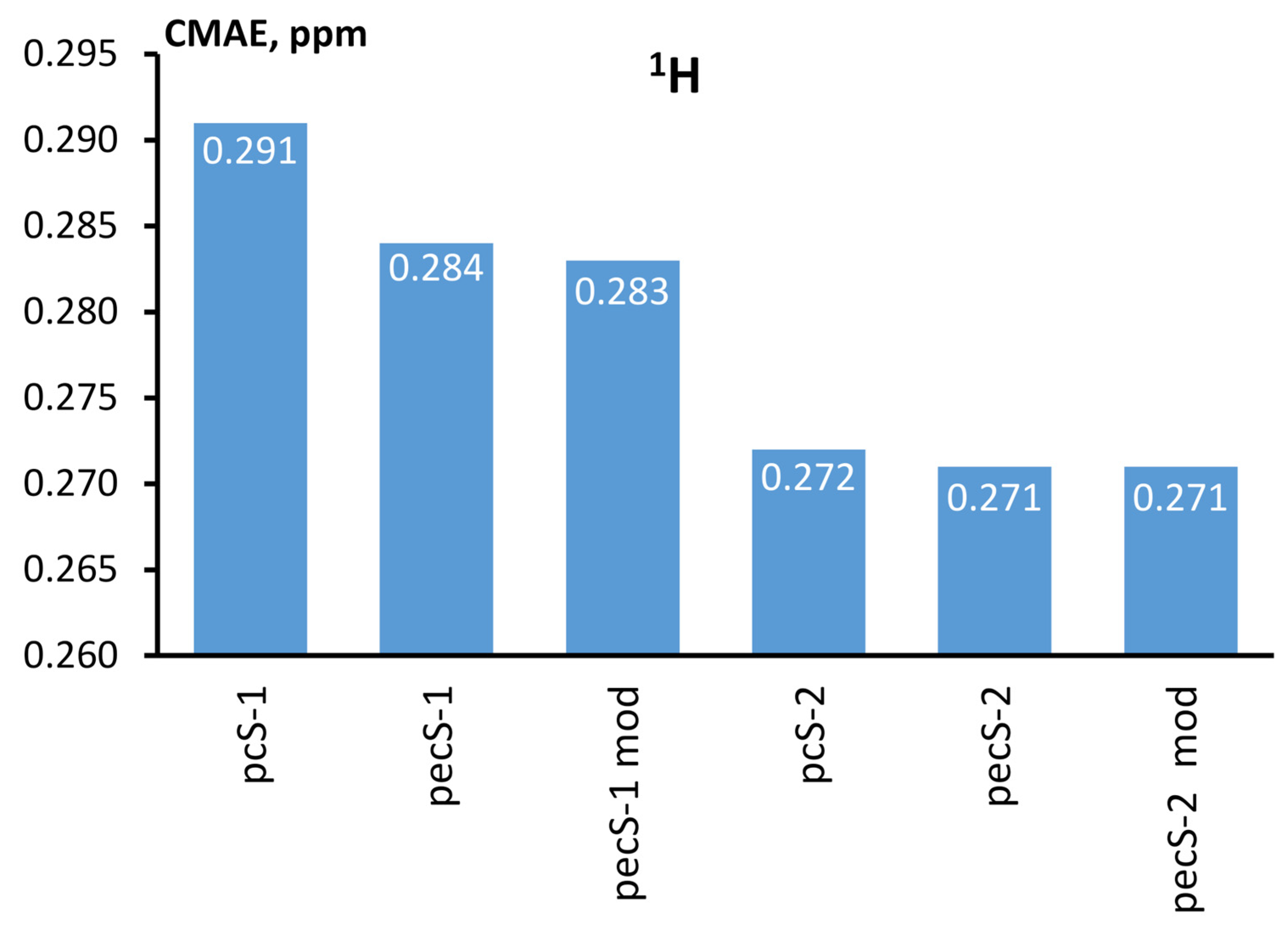{kind=link}
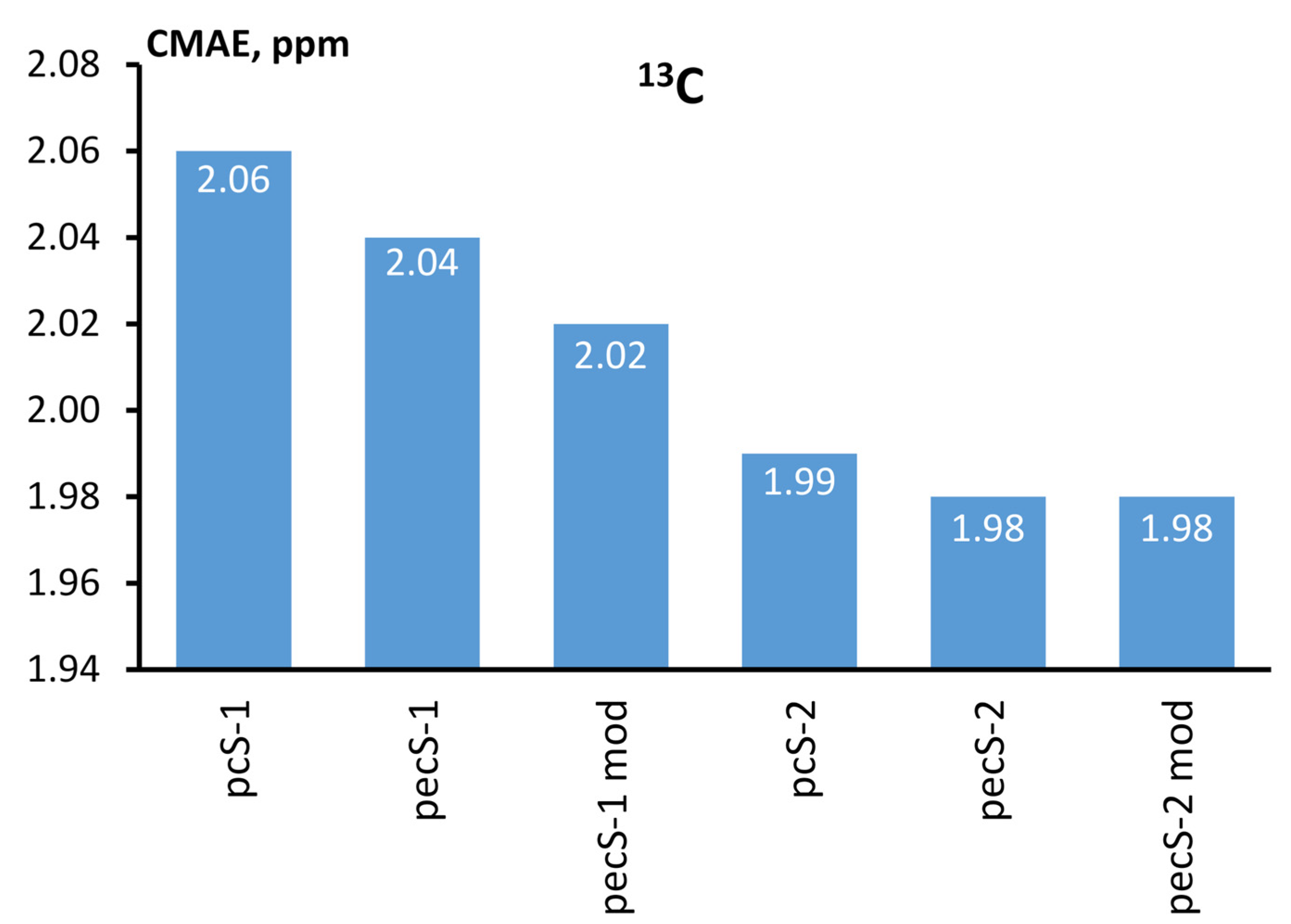{kind=link}
{kind=link}
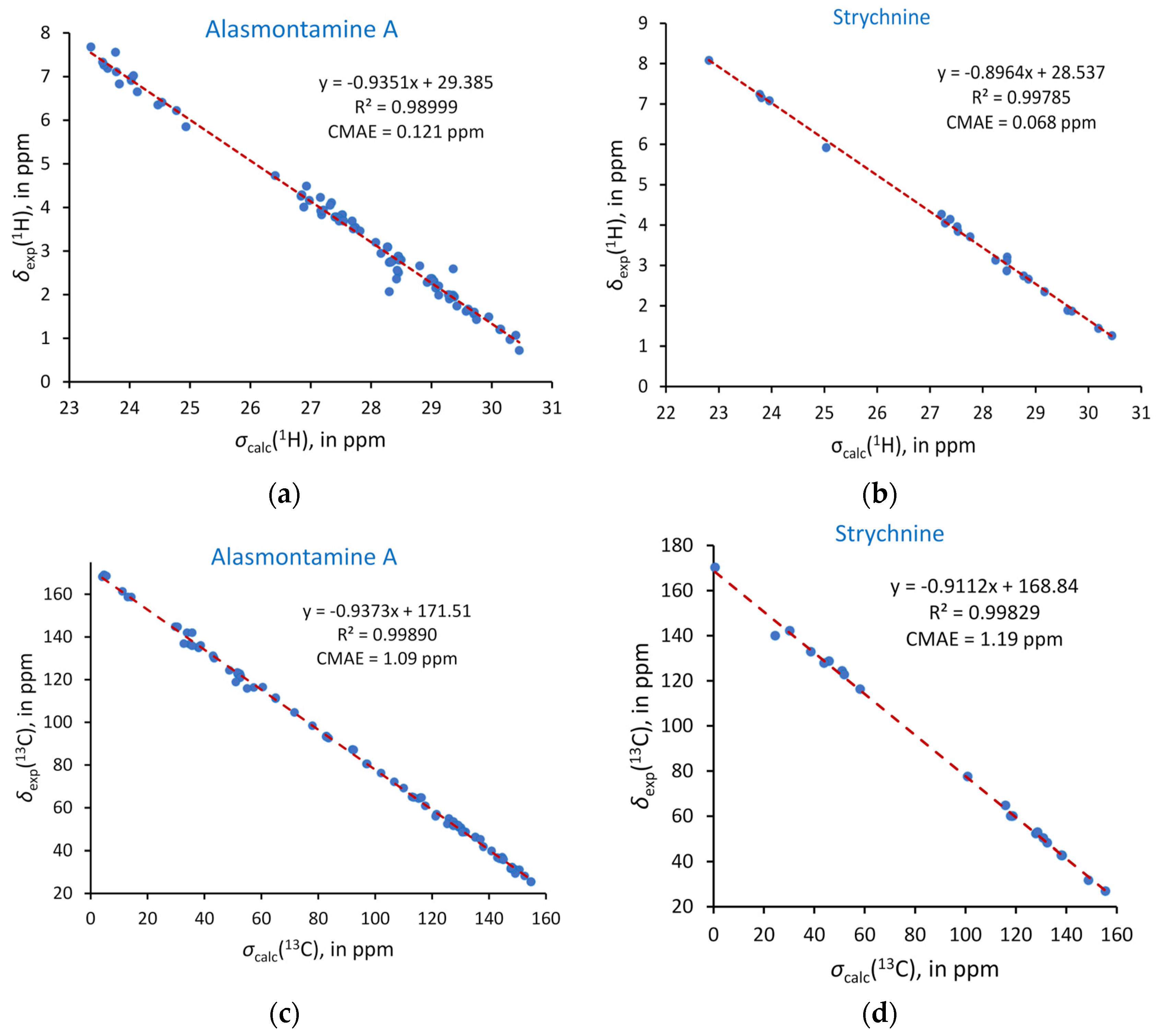{kind=link}
{kind=link}
| Atom | Basis Set | [Uncontracted| Contracted] Composition | Detailed Contraction of the p-Shell | Number of Uncontracted Basis Set Functions, Nuc | Number of Contracted Basis Set Functions, Nc | Cont. Depth (Nuc − Nc)/Nuc, % | MACE, in ppm | MAPCE, in % |
|---|---|---|---|---|---|---|---|---|
| H | original pecS-1 | [4s2p|2s1p] | 2p → (2) | 10 | 5 | 50 | 0.03 | 0.1 |
| modified pecS-1 | [4s2p|2s2p] | 2p → (1, 1) | 10 | 8 | 20 | 0.03 | 0.1 | |
| original pecS-2 | [6s3p1d|3s2p1d] | 3p → (2, 1) | 20 | 14 | 30 | 0.03 | 0.1 | |
| modified pecS-2 | [6s3p1d|3s3p1d] | 3p → (1, 1, 1) | 20 | 17 | 15 | 0.01 | 0.0 | |
| C | original pecS-1 | [7s5p1d|4s3p1d] | 5p → (3, 1, 1) | 27 | 18 | 33 | 0.39 | 0.5 |
| modified pecS-1 | [7s5p1d|4s4p1d] | 5p → (2, 1, 1, 1) | 27 | 21 | 22 | 0.18 | 0.3 | |
| original pecS-2 | [10s7p2d1f|5s4p2d1f] | 7p → (4, 1, 1, 1) | 48 | 34 | 29 | 0.45 | 0.6 | |
| modified pecS-2 | [10s7p2d1f|5s5p2d1f] | 7p → (3, 1, 1, 1, 1) | 48 | 37 | 23 | 0.05 | 0.0 |
| 1H NMR | 13C NMR | |||||
|---|---|---|---|---|---|---|
| Basis Set | A | B | R2 | A | B | R2 |
| pcS-1 | −0.92079 | 29.32047 | 0.96457 | −0.95991 | 179.6763 | 0.99657 |
| pecS-1 | −0.94085 | 30.03978 | 0.96750 | −0.93108 | 173.1887 | 0.99660 |
| pecS-1 mod | −0.93398 | 29.88718 | 0.96765 | −0.94531 | 175.7673 | 0.99663 |
| pcS-2 | −0.90872 | 28.87741 | 0.96807 | −0.93833 | 173.0559 | 0.99674 |
| pecS-2 | −0.91012 | 28.87941 | 0.96850 | −0.93670 | 172.2379 | 0.99676 |
| pecS-2 mod | −0.91125 | 28.94657 | 0.96860 | −0.94024 | 172.9650 | 0.99676 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusakov, Y.Y.; Semenov, V.A.; Rusakova, I.L. On the Efficiency of the Density Functional Theory (DFT)-Based Computational Protocol for 1H and 13C Nuclear Magnetic Resonance (NMR) Chemical Shifts of Natural Products: Studying the Accuracy of the pecS-n (n = 1, 2) Basis Sets. Int. J. Mol. Sci. 2023, 24, 14623. https://doi.org/10.3390/ijms241914623
Rusakov YY, Semenov VA, Rusakova IL. On the Efficiency of the Density Functional Theory (DFT)-Based Computational Protocol for 1H and 13C Nuclear Magnetic Resonance (NMR) Chemical Shifts of Natural Products: Studying the Accuracy of the pecS-n (n = 1, 2) Basis Sets. International Journal of Molecular Sciences. 2023; 24(19):14623. https://doi.org/10.3390/ijms241914623
Chicago/Turabian StyleRusakov, Yuriy Yu., Valentin A. Semenov, and Irina L. Rusakova. 2023. "On the Efficiency of the Density Functional Theory (DFT)-Based Computational Protocol for 1H and 13C Nuclear Magnetic Resonance (NMR) Chemical Shifts of Natural Products: Studying the Accuracy of the pecS-n (n = 1, 2) Basis Sets" International Journal of Molecular Sciences 24, no. 19: 14623. https://doi.org/10.3390/ijms241914623