
Abstract
Six new triterpenoid saponins, aesculusosides A–F (1–6), together with 19 known ones, were isolated from the seeds of Aesculus chinensis. The new structures were elucidated through extensive spectroscopic analyses and by comparison with previously reported data. Some of the isolates were evaluated for their cytotoxic activities against MCF-7 cell line by an MTT assay, and compounds 15, 16, 19, and 23–25 exhibited inhibitory activities against MCF-7 with IC50 values ranging from 7.1 to 31.3 μM.
Graphical Abstract

Similar content being viewed by others


1 Introduction
Hippocastanaceae is known to be a rich source of escins, a group of structurally diverse natural products characterized by a pentacyclic triterpenoid framework combined with a oligoglycoside chain [1]. Modern pharmacological studies show that some of these triterpenoid saponins possess diverse activities, including anti-inflammatory [2, 3], antitumor [4,5,6,7], antiviral [8], antioxidative [9, 10] and antigenotoxic properties [10].
Aesculus chinensis Bge. (Hippocastanaceae), abundant in the northwestern China, is a medicinal plant and its dried ripe seeds have been used as a stomachic and analgesic in the treatment of ditension and pain in the chest and abdomen [8]. Previous investigations on the chemical constituents of the seeds led to the isolation of an array of triterpenoid saponins [2, 8, 11,12,13]. In order to search for bioactive constituents from natural sources, we conducted the phytochemical investigation on the seeds of A. chinensis and identified six new (1–6) and 19 known triterpenoid saponins (7–25) (Fig. 1). Reported herein are the isolation and structure elucidation of compounds 1–6, as well as the cytotoxicities of some isolates.

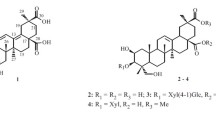
Structures of compounds 1–25
2 Results and Discussion
Compound 1 was isolated as an amorphous powder, with the molecular formula of C50H80O23 as determined by its 13C-NMR data and negative-ion HR-ESI–MS (m/z 1047.5035 ([M−H]−), calc. 1047.5018). The IR absorption bands at 3424 and 1721 nm−1 implied the presence of the hydroxyls and carboxyl groups, respectively. In the 1H-NMR spectrum (Table 1), it displayed the occurrence of one olefinic proton (δH 5.28) and six methyls (δH 1.43, 1.21, 0.96, 0.93, 0.92, and 0.88), which was in conformity with the appearance of one olefinic C-atom (δC 129.9) and six methyls (δC 30.3, 27.6, 23.0, 19.2, 17.5, and 16.4) in its 13C-NMR data, characteristic of a triterpenoid skeleton [15]. In addition, the presence of three anomeric carbon signals at δC 104.9, 104.8 and 104.2, as well as other oxygenated carbon signals in the region of δC 83.0–61.9 in the 13C-NMR spectrum, indicated that there existed a trisaccharide moiety. The above evidence suggested that the structure of 1 showed a close resemblance to that of aesculuside-B [15], except for the presence of an additional acetyl group. Careful analysis of its HSQC and 1H-1H COSY spectra revealed the presence of the following fragments, a (C-1/C-2/C-3), b (C-15/C-16), c (C-21/C-22), d (C-1′/C-2′/C-3′/C-4′), e (C-1″/C-2″/C-3″/C-4″/C-5″/C-6″), and f (C-1′′′/C-2′′′/C-3′′′/C-4′′′/C-5′′′/C-6′′′) as shown in Fig. 2. As observed in the HMBC spectrum, a correlation between the signals of CH2 (δH 3.84 and 3.76, H2-28) and the acetyl carbons (δC 173.0 and 21.0) revealed that the acyl group was attached to C-28. Furthermore, the trisaccharidic structure was characterized by HMBC cross-peaks of H-1′ (δH 4.50)/C-3 (δC 92.5), H-1″ (δH 4.83)/C-2′ (δC 80.2) and H-1′′′ (δH 4.42)/C-4′ (δC 82.9). In the ROESY spectrum of 1, there were no solid correlations which can be used to establish the relative configurations of C-3, C-16, C-21, and C-22. Thus, alkaline hydrolysis of 1 with 1% NaOMe, which liberated aesculuside-B, was conducted to assign the stereochemistry of chiral centers in 1 [15]. Furthermore, the coupling constant of H-21 and H-22 (J = 9.6 Hz) also supported the proposed stereochemistry of C-21 and C-22 [8]. Acid hydrolysis of 1 yielded glucose and glucuronic acid. The three monosaccharides were determined to be one β-d-glucuronopyranosyl acid and two β-d-glucopyranoses inferring from the coupling constants of the anomeric protons (δH 4.50, d, J = 7.6 Hz, H-1′; δH 4.83, d, J = 7.8 Hz, H-1″) and the typical carbon chemical shifts (δC 104.9, C-1′; δC 104.2, C-1″; δC 104.7, C-1′′′). Based on the above evidence, compound 1 was characterized as 28-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid.

Key COSY and HMBC correlations of compounds 1–6
Compound 2, obtained as an amorphous powder, was assigned to have a molecular formula of C52H82O24 by negative-ion HR-ESI–MS (m/z 1089.5107 ([M-H]−), calc. 1089.5123). The 1H and 13C NMR data of 2 (Tables 1, 2) exhibited an identical trisaccharide moiety and aglycone as 1. The only difference between them was an additional acetyl group in 2, with an extra characteristic proton signal at δH 2.08 in its 1H-NMR spectrum and two additional carbon signals at δC 173.5, 21.4 in its 13C-NMR spectrum. Detailed analysis of its 1D and 2D NMR spectra suggested that the additional acetyl group was attached to C-22, which could be deduced by the key HMBC cross-peak of H-22 (δH 5.21)/C-1″″ (δC 173.5). The downfield shift of H-22 further supported the above conclusion. Therefore, 2 was established as 22,28-O-diacetyl-protoaescigenin 3-O-[β-d-glucopyranosyl-(1→2)][β-d-glucopyranosyl-(1→4)]-β-d-glucuronopyranosyl acid.
Compound 3 was determined to have the same molecular formula of C50H80O23 as 1 by its negative-ion HR-ESI–MS (m/z 1047.5005 ([M−H]−), calc. 1047.5018). The characteristic NMR data suggested that 3 had the similar structure as 1. The significant differences in 1H and 13C-NMR spectra from those of 1 and 3 were chemical shifts of C-21(δC 82.8) and C-22 (δC 73.9) with corresponding protons at δH 5.50 and 3.97. The downfield shifts of C-21 implied that the acetyl group at the C-22 in 1 was transferred to C-21 in 3, which was further confirmed by the key HMBC cross-peak of H-21(δH 5.50)/C-1″″ (δC 174.0). In addition, the alkaline hydrolysis of 3 yielded aesculuside-B [15]. Hence, the structure of 3 was determined as 21-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid.
The HR-ESI–MS data of 4 showed a [M−H]− ion at m/z 1117.5425 (calc. 1117.5436), corresponding to a molecular formula of C54H86O24. The similar NMR data between 4 and 2 indicated that they are very close in the structure. Furthermore, the 1H-NMR spectrum of 4 displayed one methine proton (δH 2.62) and two doublet methyl protons (δH 1.18 and 1.19), with the corresponding carbon resonances at δC 35.7, 19.5 and 19.9 in its 13C-NMR spectrum. An isopropyl group was easily assembled by 1H-1H COSY correlations of H-2″″ (δH 2.62) with H-3″″ (δH 1.18) and H-4″″ (δH 1.19). The aforementioned data, together with the key HMBC cross-peaks of H-22 (δH 5.23)/C-1″″ (δC 179.3), H-2″″ (δH 2.62)/C-1″″ (δC 179.3), H-3″″ (δH 1.18)/C-1″″ (δC 179.3) and H-4″″ (δH 1.19)/C-1″″ (δC 179.3), suggested that the isopropyl group in 4 was installed at the C-1″″. Based on the above evidence, compound 4 was characterized as 22-O-isobutyryl-28-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid.
Compound 5 was isolated as an amorphous powder. Its molecular formula was determined by negative HR-ESI–MS [m/z 1075.5406 (calc. 1075.5331)] as C52H84O23, requiring 11 degrees of unsaturation. Careful comparison of the 1H and 13C-NMR spectra of 5 with those of 4 indicated a high degree of similarity. The differences between 5 and 4 were the absence of an acetic group at the C-28 and that the isopropyl group was transferred to C-21 other than C-22 in compound 4, as confirmed by the combinational analysis of 2D NMR data (Fig. 2). Thus, compound 5 was elucidated as 21-O-isobutyrylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid.
Compound 6 was obtained as an amorphous powder and gave a molecular formula of C52H82O23, as determined by HR-ESI–MS at m/z 1073.5162 (calc. 1073.5174). The 1H-NMR spectrum of 6 (Table 1) revealed nine methyls [δH 0.84 (3H, s), 0.86 (3H, s), 0.93 (3H, s), 0.97 (3H, s), 1.00 (3H, s), 1.09 (3H, s), 1.44 (3H, s), 2.06 (3H, s), 2.09 (3H, s)] and one olefinic proton (δH 5.31).The above data, together with its 13C-NMR data (Table 2), indicated that aglycone moiety in 6 was barringtogenol C [16]. Furthermore, the characteristic carbon resonances displayed in the region of δC 83.0–62.6 in its 13C-NMR data (Table 2), indicated that there also existed a trisaccharide moiety. Acid hydrolysis of 6 also yielded glucose and glucuronic acid. The HMBC correlations from H-21 to C-20/C-22/C-29/C-30/C-1″″, and from H-28 to C-1″″, allowed for the assignment of two acetyl groups as shown in Fig. 1. Hence, the structure of 6 was deduced as 21,28-O-diacetylbarringtogenol C 3-O-[β-d-glucopyranosyl (1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid.
The 19 known compounds were identified as 22-O-tigloylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Escin Ivg] (7) [17], 21,22-O-diacetylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Escin Iv] (8) [18], 22-O-angeloylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Escin Ivh] (9) [2], 21,28-O-diacetylprotoaescigenin 3-O-[β-d-glucopyranosyl (1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Aesculiside A] (10) [19], 22-O-tigloyl-28-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Escin IVc] (11) [20], 22-O-angeloyl-28-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl (1→2)] [β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Escin IVd] (12) [20], 21-O-tigloylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Aesculioside A] (13) [13], 21-O-angeloylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Aesculioside B] (14) [13], 21-O-isobutyryl-22-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Escin V] (15) [18], 21-O-tigloyl-22-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl (1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid (Escin Ia) (16) [20], 21-O-isobutyryl-28-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl(1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Isoescin V] (17) [18], 21-O-angeloyl-22-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl (1→2)] [β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid (Escin Ib) (18) [21], 21-O-isovaleryl-22-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl (1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Esvin VI] (19) [18], 21-O-tigloyl-28-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl (1→2)] [β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid (isoescin Ia) (20) [20], 21-O-angeloyl-28-O-acetylprotoaescigenin 3-O-[β-d-glucopyranosyl (1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid (isoescin Ib) (21) [20], 21-O-tigloyl-22-O-acetylprotoaescigenin 3-O-[β-d-galactopyranosyl(1→2)] [β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid (22) [22], 21-O-tigloyl-22-O-acetylbarringtogenol C 3-O-[β-d-glucopyranosyl (1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Escin IIIa] (23) [21], 21-O-tigloyl-28-O-acetylbarringtogenol C 3-O-[β-d-glucopyranosyl (1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Isoescin IIIa] (24) [21], 21-O-angeloyl-28-O-acetylbarringtogenol C 3-O-[β-d-glucopyranosyl (1→2)][β-d-glucopyranosyl(1→4)]-β-d-glucopyranosiduronic acid [Isoescin IIIb] (25) [11].
The known compounds 7-9, 12-17, 19, and 22–25 were evaluated for their in vitro cytotoxicities against human cancer cell line (MCF-7) using the MTT method as reported previously [14], with cis-platin as positive control (Table 3). Compound 23 showed significant toxicity effect against MCF-7 with IC50 value of 7.1 μM, while 15 and 24 showed moderate cytotoxicities against MCF-7 cell line, with IC50 values of 16.9 and 11.8 μM, respectively.
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were measured with Perkin Elmer/Model-343 digital polarimeter. IR spectra were recorded on a JASCO FT/IR-480 spectrophotometer and reported as wave number (cm−1). 1H, 13C NMR spectra and 2D NMR spectra were recorded on a Bruker Avance III 600 spectrometer. Chemical shifts were reported using TMS as the internal standard. HR-ESI–MS data were obtained on a Bruker Apex IV FT-MS spectrometer. Column chromatography (CC) was carried out using D-101 macroreticular resin (Tianjin Polymer Technology Co. Ltd.), MCI gel (75–150 μm, Mitsubishi Chemical Industries, Japan) and silica gel (90–200 µm; Qingdao Marine Chemical Co. Ltd., Qingdao, People’s Republic of China). MPLC was performed on a Lisui EZ Purify III System packed with RP-18 silica gel (40–63 μm, Merck, 71 Darmstadt, Germany) columns. Precoated silica gel GF254 plates (Qingdao Marine Chemical Co. Ltd, Qingdao, People’s Republic of China) were used for thin-layer chromatography (TLC). Preparative HPLC was performed on Shimadzu LC-8A equipped with a Shimadzu PRC-ODS(K) column and Agilent 1100 apparatus equipped with a Zorbax SB-C-1875 (Agilent, 9.4 mm × 25 cm) column, respectively.
3.2 Plant Material
The seeds of A. chinensis were collected from Enshi, Hubei Province, P. R. China in October 2014, and were identified by Dr. Wei Sun (Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences). A voucher specimen (201410 M) was deposited in the herbarium at the department of medicinal plants, Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences (Beijing 100700, China).
3.3 Extraction and Isolation
The dried seeds of A. chinensis (10 kg) were extracted with 70% ethanol under reflux for three times (3, 2, and 1 h, respectively). The resultant extract was resolved in H2O and extracted with EtOAc for three times. After removal of the EtOAc fraction, the remaining solution was then extracted with butanol for three times. The butanol solution was concentrated under reduced pressure and the butanol fraction (56 g) was subjected to D-101 (eluted with 20% ethanol, 40% ethanol, 70% ethanol and 95% ethanol) to afford four fractions. The 40% ethanol-eluted fraction (12 g) was decolorized over MCI gel (eluted with 90% MeOH) and then was subjected to MPLC (MeOH/H2O 5–35%) to provide Frs. 1–5. Frs. 1 (4.8 g) was further separated by using repeated prep. HPLC (MeOH/H2O 8–20%) to give five subfractions, Frs. 1A-1E. Subfraction Frs. 1A (0.4 g) was purified by semipreparative HPLC (MeCN/H2O, 11%) to afford compounds 1 (11 mg), 7 (8 mg), 8 (23 mg), and 9 (4 mg). Compounds 2 (5 mg), 3 (4 mg), 10 (16 mg), 11 (5 mg), 12 (7 mg), and 13 (8 mg) were obtained by two times of semipreparative HPLC (MeCN/H2O, 13%) from Frs. 1B (0.6 g). Subfraction Frs. 1C (0.1 g) was separated by semipreparative HPLC (MeOH/H2O, 15%) to afford 4 (2 mg) and 5 (1 mg). Subfraction Frs. 1D (0.3 g) was subjected to semipreparative HPLC (MeOH/H2O, 18%) to give compounds 6 (8 mg), 14 (5 mg), 15 (2 mg), and 22 (4 mg). Compounds 16 (6 mg), 17 (11 mg), and 18 (15 mg) were obtained from Frs. 2 by repeated prep. HPLC purification. Frs. 3 was subjected to repeated prep. HPLC and semi-perp. HPLC to afford compounds 19 (5 mg) and 23 (2 mg). From Frs. 4 and Frs. 5, compounds 20 (4 mg), 21 (4 mg), 24 (3 mg) and 25 (1 mg) were obtained by repeated prep. HPLC and semi-perp. HPLC.
3.3.1 Aesculusoside A (1)
Amorphous powder; [α] 25D − 2.5 (c 0.1, MeOH); IR (KBr) 3423, 2951, 1721, 1380, 1261, 1031 cm−1; 1H and 13C NMR data: see Tables 1 and 2; HR-ESI–MS (m/z 1047.5035 ([M−H]−), calc. 1047.5018).
3.3.2 Aesculusoside B (2)
Amorphous powder; [α] 25D − 3.5 (c 0.1, MeOH); IR (KBr) 3421, 2945, 1725, 1384, 1272, 1077 cm−1; 1H and 13C NMR data: see Tables 1 and 2; HR-ESI–MS (m/z 1089.5107 ([M−H]−), calc. 1089.5123).
3.3.3 Aesculusoside C (3)
Amorphous powder; [α] 25D − 34.5 (c 0.1, MeOH); IR (KBr) 3422, 2928, 1720, 1383, 1273, 1076 cm−1; 1H and 13C NMR data: see Tables 1 and 2; HR-ESI–MS (m/z 1047.5005 ([M−H]−), calc. 1047.5018).
3.3.4 Aesculusoside D (4)
Amorphous powder; [α] 25D − 32.1 (c 0.1, MeOH); IR (KBr) 3421, 2946, 1723, 1383, 1277, 1074 cm−1; 1H and 13C NMR data: see Tables 1 and 2; HR-ESI–MS (m/z 1117.5425 ([M−H]−), (calc. 1117.5436).
3.3.5 Aesculusoside E (5)
Amorphous powder; [α] 25D − 2.5 (c 0.1, MeOH);) IR (KBr) 3423, 2930, 1721, 1375, 1277, 1075 cm−1; 1H and 13C NMR data: see Tables 1 and 2; HR-ESI–MS (m/z 1075.5406 ([M−H]−), (calc. 1075.5331).
3.3.6 Aesculusoside F (6)
Amorphous powder; [α] 25D − 2.8 (c 0.1, MeOH); IR (KBr) 3423, 2929, 1721, 1384, 1273, 1075 cm−1; 1H and 13C NMR data: see Tables 1 and 2; HR-ESI–MS (1073.5162 ([M−H]−), (calc. 1073.5174).
3.4 Cytotoxicity Assay
Compounds were tested in vitro for their cytotoxicities against proliferation of MCF-7 (breast cancer) using the MTT method [14]. The human tumor cell line MCF-7 was obtained from ATCC (Manassas, VA, USA). All cells were cultured in DMEM medium (Biological Industries, Kibbutz Beit-Haemek, Israel), which were supplemented with 10% fetal bovine serum (Biological Industries, Kibbutz Beit-Haemek, Israel) at 37 °C in a humidified atmosphere containing 5% CO2. Briefly, cells were seeded into each well of a 96-well cell culture plate. After 12 h of incubation at 37 °C, the test compound (40 μM) was added. After incubation for 48 h, cells were subjected to the MTT assay. Compounds with a growth inhibition rate of 50% were further evaluated with cis-platin (Sigma, St. Louis, MO, USA) as positive control.
3.5 Acid Hydrolysis of Compounds 1 and 6
A solution of compound 1 (4 mg) in H2O (1 mL) was treated with 20% aqueous H2SO4 (1 mL), and the mixture was heated under reflux for 2 h. It was then neutralized by saturated NaHCO3 and extracted three times with EtOAc. Glucose and glucuronic acid were obtained from the H2O layer, and identified by comparison with authentic samples and by PC (Paper Chromatography) behavior, solvent: CHCl3-MeOH-H2O, 10:6:1.
3.6 Alkaline Hydrolysis of Compound 1
Compound 1 (5 mg) was added to a MeOH solution (5 ml) of NaOMe (1 mg). The mixture was stirred at room temperature for 4 h and then neutralized with 20% aqueous HCl. The reaction mixture was concentrated under reduced pressure, and the residue was purified by column chromatography on silica gel (CH2Cl2/MeOH 2:1–1:1) to furnish aesculuside-B (1.2 mg, 46% yield), which was identical with authentic sample by TLC, 1H- and 13C-NMR spectra comparisons [15].
References
Z. Zhang, S. Li, X.Y. Lian, Crops 1, 24–51 (2010)
F. Wei, L.Y. Ma, W.T. Jin, S.C. Ma, G.Z. Han, I.A. Khan, R.C. Lin, Chem. Pharm. Bull. 52, 1246–1248 (2004)
H. Matsuda, Y. Li, T. Murakami, K. Ninomiya, J. Yamahara, M. Yoshikawa, Biol. Pharm. Bull. 20, 1092–1095 (1997)
X.Y. Zhou, F.H. Fu, Z. Li, Q.J. Dong, J. He, C.H. Wang, Planta Med. 75, 1580–1585 (2009)
Y.P. Niu, L.D. Li, L.M. Wu, Lymphoma 49, 1384–1391 (2008)
Y.P. Niu, L.M. Wu, Y.L. Jiang, W.X. Wang, L.D. Li, J. Pharm. Pharmacol. 60, 1213–1220 (2008)
J.M.R. Patlolla, J. Raju, M.V. Swamy, C.V. Rao, Mol. Cancer Ther. 5, 1459–1466 (2006)
X.W. Yang, J. Zhao, Y.X. Cui, X.H. Liu, C.M. Ma, M. Hattori, L.H. Zhang, J. Nat. Prod. 62, 1510–1513 (1999)
I. Kucukkurt, S. Ince, H. Keles, E.K. Akkol, G. Avci, E. Yesilada, E. Bacak, J. Ethnopharmacol. 129, 18–22 (2010)
I. Sato, T. Suzuki, H. Kobayashi, S. Tsuda, J. Vet. Med. Sci. 67, 731–734 (2005)
J. Zhao, X.W. Yang, J. Asian Nat. Prod. Res. 5, 197–203 (2003)
J. Zhao, X.W. Yang, M. Hattori, Chem. Pharm. Bull. 49, 626–628 (2001)
Z. Zhang, K. Koike, Z. Jia, T. Nikaido, D. Guo, J. Zheng, Chem. Pharm. Bull. 47, 1515–1520 (1999)
T. Mosmann, J. Immunol. Methods 65, 55–63 (1983)
B. Singh, P.K. Agrawal, R.S. Thakur, J. Nat. Prod. 50, 781–783 (1987)
M. Yoshikawa, E. Harada, T. Murakami, H. Matsuda, N. Wariishi, J. Yamahara, N. Murakami, I. Kitagawa, Chem. Pharm. Bull. 42, 1357–1359 (1994)
X.W. Yang, J. Zhao, Y.X. Cui, Chin. Chem. Lett. 11, 139–142 (2000)
M. Yoshikawa, T. Murakami, J. Yamahara, H. Matsuda, Chem. Pharm. Bull. 46, 1764–1769 (1998)
X.W. Yang, J. Guo, Zhongguo Xinyao Zazhi 16, 1373–1376 (2007)
J. Guo, X.W. Yang, J. Chin. Pharm. Sci. 13, 87–91 (2004)
X. Yang, J. Zhao, S. Ouyang, Zhongcaoyao 33, 389–391 (2002)
F. Wei, L. Ma, S. Ma, R. Lin, Yaowu Fenxi Zazhi 24, 400–402 (2004)
Acknowledgements
We gratefully acknowledge financial support from the independent topics supported by operational expenses for basic research of China Academy of Chinese Medical Sciences (ZXKT15032).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cheng, JT., Chen, ST., Guo, C. et al. Triterpenoid Saponins from the Seeds of Aesculus chinensis and Their Cytotoxicities. Nat. Prod. Bioprospect. 8, 47–56 (2018). https://doi.org/10.1007/s13659-017-0148-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-017-0148-4