Comparative and Phylogenetic Analysis of Complete Chloroplast Genomes in Eragrostideae (Chloridoideae, Poaceae)




Shandong Provincial Key Laboratory of Plant Stress Research, College of Life Science, Shandong Normal University, No. 88 Wenhuadong Road, Jinan 250014, China
*
Authors to whom correspondence should be addressed.
Plants 2021, 10(1), 109; https://doi.org/10.3390/plants10010109
Submission received: 3 December 2020
/
Revised: 3 January 2021
/
Accepted: 4 January 2021
/
Published: 6 January 2021
(This article belongs to the Special Issue Plant Evolution, Systematics, and Chloroplast Genome)
Abstract
:Eragrostideae Stapf, the second-largest tribe in Chloridoideae (Poaceae), is a taxonomically complex tribe. In this study, chloroplast genomes of 13 Eragrostideae species were newly sequenced and used to resolve the phylogenetic relationships within Eragrostideae. Including seven reported chloroplast genomes from Eragrostideae, the genome structure, number and type of genes, codon usage, and repeat sequences of 20 Eragrostideae species were analyzed. The length of these chloroplast genomes varied from 130,773 bp to 135,322 bp. These chloroplast genomes showed a typical quadripartite structure, including a large single-copy region (77,993–80,643 bp), a small single-copy region (12,410–12,668 bp), and a pair of inverted repeats region (19,394–21,074 bp). There were, in total, 129–133 genes annotated in the genome, including 83–87 protein-coding genes, eight rRNA genes, and 38 tRNA genes. Forward and palindromic repeats were the most common repeat types. In total, 10 hypervariable regions (rpl22, rpoA, ndhF, matK, trnG–UCC-trnT–GGU, ndhF–rpl32, ycf4–cemA, rpl32–trnL–UAG, trnG–GCC–trnfM–CAU, and ccsA–ndhD) were found, which can be used as candidate molecular markers for Eragrostideae. Phylogenomic studies concluded that Enneapogon diverged first, and Eragrostis including Harpachne is the sister to Uniola. Furthermore, Harpachne harpachnoides is considered as a species of Eragrostis based on morphological and molecular evidence. In addition, the interspecies relationships within Eragrostis are resolved based on complete chloroplast genomes. This study provides useful chloroplast genomic information for further phylogenetic analysis of Eragrostideae.
1. Introduction
Chloroplasts are the organelles necessary for photosynthesis, and the most important and common plastids in plant cells. In addition, chloroplasts are semi-autonomous organelles with their own genome, which is a relatively independent genetic system in plant cells. Compared with the nuclear genome, the chloroplast genome has the characteristics of small size, single parent inheritance, low nucleotide substitution rate, and highly conserved genome structure. In angiosperms, the chloroplast genome is relatively conservative, and has a typical quadripartite structure with a pair of inverted repeat regions (IRb/IRa), a large single copy (LSC) region, and a small single copy (SSC) region [1]. The length of the chloroplast genome varies greatly from species to species [2]. With the rapid development of next-generation sequencing (NGS) technology, it is easier to obtain the complete chloroplast genome, making chloroplast genomes a research hotspot [3,4,5]. The chloroplast genome has important value in studying the phylogeny of species [6,7]. The research scope of chloroplast genomes is also relatively wide, including comparative genomics research, phylogenetic research, and simple sequence repeat (SSR) genetic polymorphism research [8,9].
Due to the limited taxonomic traits that can be reflected by herbarium specimens, Chloridoideae has always been one of the most difficult groups to study in Poaceae systematics [10,11]. Peterson et al., (2010) [12] divided Chloridoideae into four tribes (Triraphideae, Eragrostideae, Zoysieae, and Cynodonteae) based on multiple gene sequences. In Eragrostideae, Cotteinae (including Cottea and Enneapogon) diverged first, and Eragrostidinae (including Ectrosia, Harpachne, Psammagrostis, and Eragrostis) is the sister to Uniolinae (including Entoplocamia, Tetrachne, and Uniola). Eragrostideae Stapf is the second-largest and more complex tribe in Chloridoideae. There are about 500 species in this tribe [13,14]. All species in Eragrostideae use the C4 photosynthetic pathway (Eragrostis walteri is a C3 plant [15,16]), and most of them are distributed in tropical and subtropical regions [14,17]. Members of Eragrostideae are generally characterized by laterally compressed spikelets, glabrous three (to 13)-nerved lemmas, and ciliate ligule [18].
Eragrostis Wolf, the type genus of Eragrostideae, is the largest genus in Eragrostideae. There are more than 400 species worldwide [19,20]. Due to its large number of species, diverse chromosome ploidy, and similar morphological characteristics between species [21], it is a complex genus in Eragrostideae. Due to its large size and wide geographical distribution, comprehensive taxonomic treatment of the genus remains difficult. Several phylogenetic studies focusing on Eragrostis and its related genera have been carried out [3,21,22,23], however, correct intergeneric and infrageneric relationships still remain unresolved. There has been some debate in the recent literature as to whether the genus is monophyletic. Several studies [3,21] suggested that Eragrostis was a monophyletic group, however, Eragrostis is considered to be a paraphyletic group in some studies [24,25]. Ingram and Doyle [23] found that Eragrostis was a monophyletic group with the inclusion of four segregate genera: Acamptoclados, Diandrochloa, Neeragrostis, and Pogonarthria based on the plastid locus rps16 and nuclear gene waxy. Peterson et al. [12] found a terminal Eragrostidinae clade of Ectrosia, Harpachne, and Psammagrostis embedded in a polyphyletic Eragrostis. In addition, the infrageneric relationships of Eragrostis could not be solved based on partial molecular sequences [21,22]. However, Somaratne et al. [3] reconstructed the relationships among the five species of Eragrostis, according to the whole chloroplast genomes. Therefore, the above shows that more evidence and broader sampling are needed to resolve the phylogenetic relationships within Eragrostis. Harpachne Hochst. was first recorded in 1841, and Harpachne harpachnoides (Hack.) B. S. Sun and S. Wang was described as “Eragrostis harpachnoides Hack.” in Oesterreichische Botanische Zeitschrift. in 1902, which shows that Harpachne is closely related to Eragrostis. Harpachne is a small genus that contains only three species: Harpachne bogdanii Kenn.-O’Byrne, Harpachne harpachnoides (Hack.) B. S. Sun and S. Wang, and Harpachne schimperi A. Rich. In Flora of China [17], Harpachne is distinguished from Eragrostis by the morphology of inflorescence, but both have ciliated ligules and three-veined lemmas. Peterson et al. [12] found that Harpachne was embedded in Eragrostis based on multi-gene phylogenetic trees, which was consistent with previous studies [13,24,25] based on a few molecular sequences. Reconstruction of the phylogenetic relationship between Eragrostis and Harpachne through complete chloroplast genomes has not been previously reported. Therefore, whether H. harpachnoides was a species of Eragrostis can be studied with both morphological and complete chloroplast genome evidence.
In this study, chloroplast genomes of 11 Eragrostis species, Enneapogon desvauxii, and Harpachne harpachnoides were newly sequenced (Table 1). A genomic comparative analysis was performed in combination with chloroplast genomes of four other Eragrostis species, one Uniola species, and two other Enneapogon species available in GenBank. In addition, we carried out anatomy investigations of the spikelets of H. harpachnoides and E. tenella, and compared their morphological difference (Figure 1). The main purpose of this study was to: (1) compare and analyze the chloroplast genome structure of the 20 Eragrostideae species; (2) identify highly divergent regions of all 20 Eragrostideae chloroplast genomes; (3) explore the phylogenetic position of Harpachne relative to Eragrostis, and resolve the interspecies relationships within Eragrostis. In summary, this study will provide important insight in understanding the chloroplast genome evolution and phylogeny of Eragrostideae species.
2. Results
2.1. Chloroplast Genome Characteristics of Eragrostideae
The complete chloroplast genome length of the 20 Eragrostideae species varied from 130,773 bp (Eragrostis tenellula) to 135,322 bp (Uniola paniculata), and showed a typical quadripartite structure with the LSC region (77,993–80,643 bp), SSC region (12,410–12,668 bp), and a pair of IR regions (19,394–21,074 bp) (Table 2). The overall guanine–cytosine (GC) content of each species was approximately 38% (Table 2). The GC content in the IR region was higher than both the LSC and SSC regions. There were 129–133 genes, including 83–87 protein-coding genes (PCGs), eight ribosomal RNA genes (rRNAs), and 38 transfer RNA genes (tRNAs) (Table 2). E. tenellula had the fewest genes, and lacked rps15. We found that there were some conserved ycf1 and ycf2 gene residues in some species of Eragrostideae, and the accD gene had completely degraded. In addition, the intron sequences of the clpP gene and the rpoC1 gene had been lost. Therefore, there were 16 intron-containing genes in each of the Eragrostideae species, of which, two PCGs (ycf3 and rps12) had two introns, and eight PCGs (ndhB, rpl2, ndhA, rpl16, petB, atpF, petD, and rps16) and six tRNAs (trnA-UGC, trnI-GAU, trnK-UUU, trnG-UCC, trnV-UAC, and trnL-UAA) had a single intron.
2.2. Repeat Sequences and SSRs Analysis
A total of 933 repeats were identified in 20 Eragrostideae chloroplast genomes through REPuter, including 578 forward repeats, 345 palindromic repeats, seven reverse repeats, and three complementary repeats. Each species detected 47 repeats on average. E. cilianensis, E. japonica, E. nigra, E. setifolia, E. tenella, and H. harpachnoides had the largest number of repeats (49), while E. pilosa had the smallest number of repeats (40; Figure 2). Three complementary repeats were only detected in the chloroplast genomes of E. japonica (one) and E. tenella (two). The length of the repeats was mainly concentrated in 30–38 bp (Figure 3).
A total of 943 SSRs were identified in the chloroplast genomes of 20 Eragrostideae species using MISA script. These SSRs were mainly distributed in the LSC region (Table 3). These SSRs were mononucleotide repeats and dinucleotide repeats, and the mononucleotide repeat type was mainly A/T repeat, while all the dinucleotide repeat sequences were composed of AT/TA repeats (Table 3). There were no trinucleotide or longer repeats in these chloroplast genomes. E. tenella had the largest number of SSRs (56), including 53 mononucleotide repeats (51 A/T repeats and two G/C repeats) and three dinucleotide repeats. E. atrovirens and E. setifolia both had the minimum number of SRRs (40). The SSRs in U. paniculata were all distributed in the LSC region. SSRs were not found in the SSC region in E. desvauxii. In these species, compound SSRs were also rich in A/T repeats, and all were located in the LSC region.
2.3. Codon Usage Analysis
By removing repeats and lengths of less than 300 bp sequences, 51 coding sequences (CDSs) were selected from 16 Eragrostideae species, and 50 CDSs were selected from four Eragrostideae species (E. japonica, E. ferruginea, E. tenellula, and U. paniculata). The analysis results showed that the variation range of the total GC content was 38.9–39.1%, with an average value of 38.97%. The result showed that the GC content was low and the difference in content between various species was not significant. The number of codons ranged from 16,999 (E. japonica) to 17,210 (E. pilosa), with an average value of 17,151 (Table 4). Among them, there were six codon types encoding leucine, serine, and arginine, while there was only one codon type encoding methionine and tryptophan (Table S1). In addition, leucine was the most amino acid encoded in the chloroplast genome, accounting for 10.82% on average of all amino acids. Cysteine had the smallest number of codons, accounting for only 1.09% on average of all amino acids (Figure 4). The values for the effective number of codons (ENC) in Eragrostideae chloroplast genomes ranged from 49.40 to 49.80, with an average value of 49.56 (Table 4). In all species, there were 31 codons with an relative synonymous codon usage (RSCU) value greater than 1.00, of which, 29 ended with A or U codons and two ended with G or C codons (UUG, UGU). In addition, the RSCU value of methionine (AUG) and tryptophan (UGG) was 1.00 (Table S2).
2.4. Expansion and Contraction of the IR Region
The expansion and contraction of the IR region in 20 Eragrostideae chloroplast genomes were analyzed via IRscope. Although the chloroplast genomes of Eragrostideae were highly conserved, there were still some differences in the IR/single cope (SC) border area (Figure 5). The IRb/SSC junction (JSB) of all Eragrostideae species (except E. oblongus) was located within the ndhF gene, and part of this gene was duplicated 21–53 bp in the IRb region. Due to the early termination of the ndhF gene in En. oblongus, JSB was located in the intergenic region between rps19 and rps15. Similarly, the SSC/IRa junction (JSA) of all Eragrostideae species (except E. oblongus and E. tenellula) was located within the ndhH gene, and this gene extended 2–4 bp into the IRa region. Unlike the above situation, JSA in E. oblongus and E. tenellula did not extend to the IRa region, so it was located in the intergenic region between ndhF and rps15. The LSC/IRb junction (JLB) was located between rpl22 and rps19. The fragment size of rpl22-rps19, located in IRb region was 30 bp in E. ferruginea, 36 bp in E. setifolia, 38 bp in E. minor, 59 bp in E. desvauxii, and 35 bp in the remaining species. The LSC/IRa junction (JLA) was located between rps19 and psbA. The fragment size of rps19-psbA located in IRa region was 30 bp in E. ferruginea, 36 bp in E. setifolia, 38 bp in E. minor, 59 bp in En. desvauxii, and 35 bp in remaining species.
2.5. Comparative Genome Analysis and Identification of Hypervariable Regions
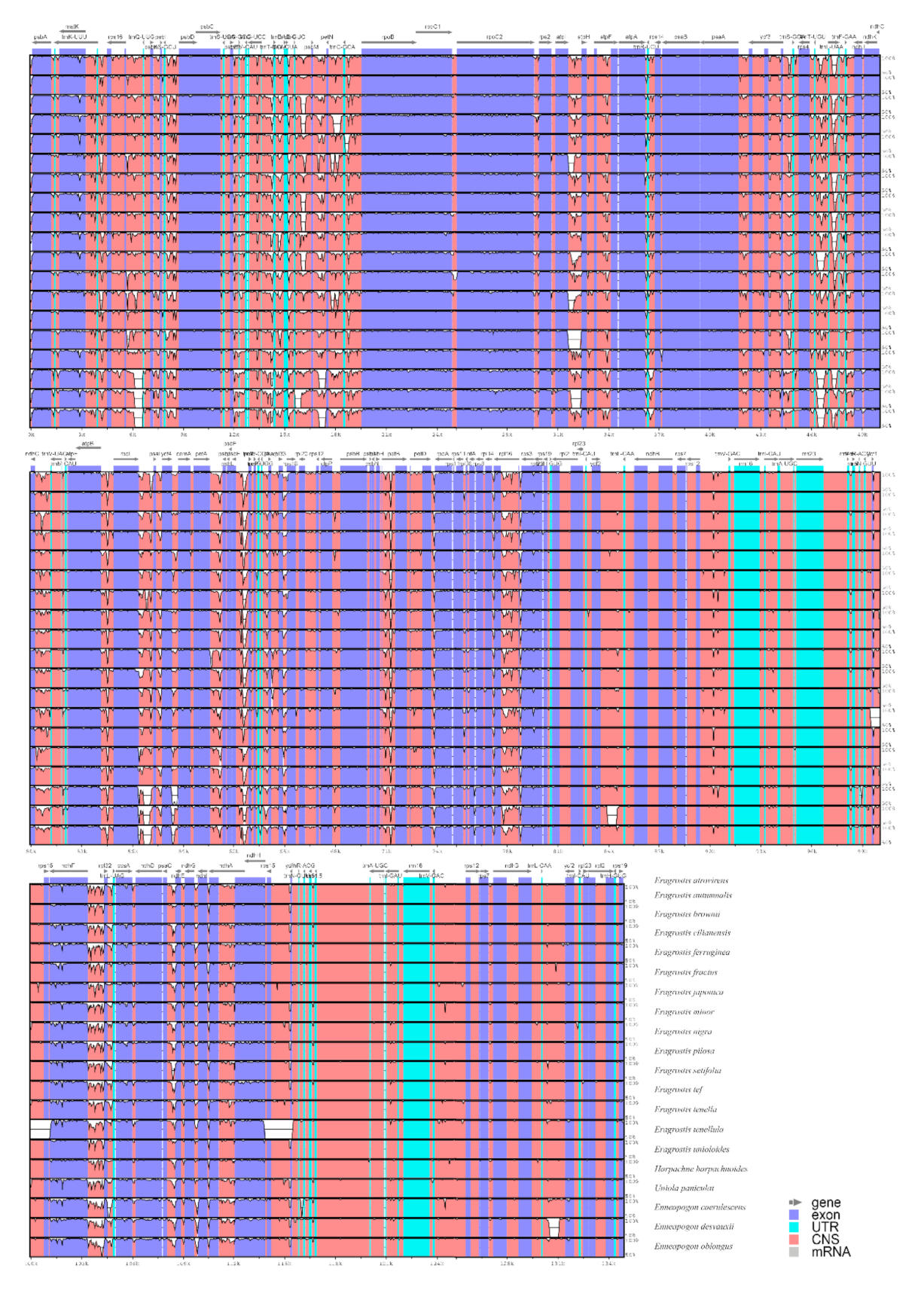
By comparing the complete chloroplast genomes, we can understand the differences in the chloroplast genome sequences between different species. In this study, the mVISTA program was used to align and compare 20 Eragrostideae chloroplast genomes with E. atrovirens as a reference (Figure 6). The results showed that all aligned chloroplast genome sequences have a high similarity. The IR regions were more conservative than the LSC region and the SSC region. The noncoding regions had a higher mutation rate than the protein-coding regions, and the intergenic spacers (IGS) were particularly prominent.
In order to further identify regions with high mutations, we performed single nucleotide polymorphism (SNP) analysis on the selected CDS and noncoding regions using MEGA v7.0.26, and counted the number of mutation sites and parsimony information sites. Then, the percentage of corresponding parsimony information sites (Pi%) was calculated. We screened 137 regions for analysis, including 58 CDS regions, 64 IGS regions, and 15 intron regions. In the 58 CDS regions, Pi% values ranged from 0.2609 (ndhB) to 5.8751 (matK). Among them, rpl22, rpoA, ndhF, and matK had significantly higher Pi% values (Pi% ≥ 4; Figure 7A). Correspondingly, in 79 noncoding regions, the Pi% values ranged from 0.1572 (trnI-CAU-ycf2) to 12.5604 (ccsA-ndhD). Six of these regions (i.e., trnG-UCC-trnT-GGU, ndhF-rpl32, ycf4-cemA, rpl32-trnL-UAG, trnG-GCC-trnfM-CAU, and ccsA-ndhD) also showed quite high Pi% values (Pi% > 8; Figure 7B). From the results, the average value of Pi% of the noncoding regions (4.8829) was about twice higher than that of the CDS regions (2.2949), indicating that the noncoding regions were more hypervariable than the CDS regions. Moreover, compared with the SC regions, the Pi% values of the IR regions were lower and relatively more conservative.
2.6. Phylogenetic Analysis of Eragrostideae
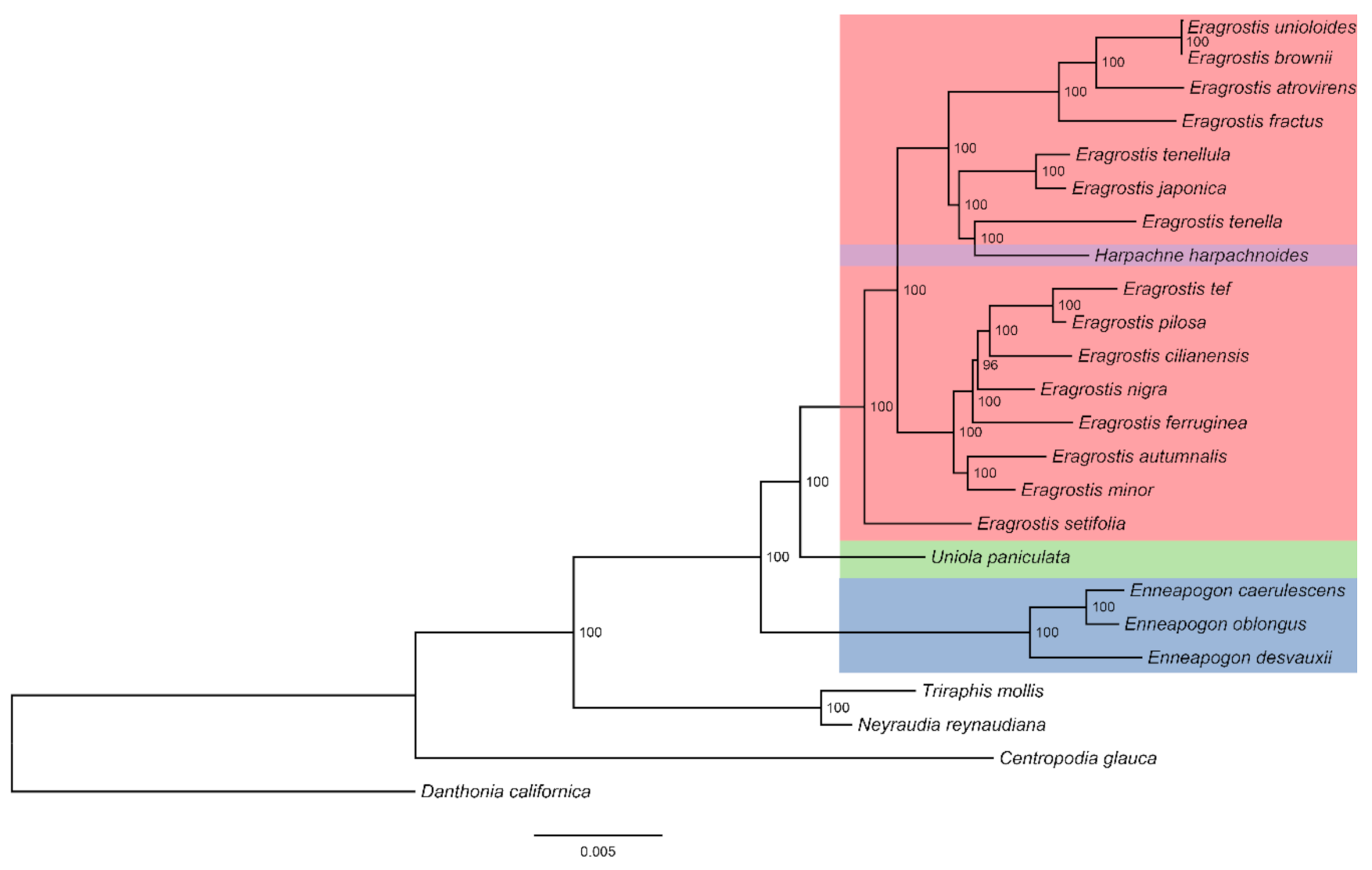
In this study, similar topologies were observed in maximum likelihood (ML) trees of seven datasets (complete chloroplast genomes, coding sequences, noncoding sequences, hypervariable regions, LSC regions, SSC regions, and IR regions) among 20 Eragrostideae species (Figure 8 and Figures S2–S7). In general, Eragrostis—including H. harpachnoides—showed a sister relationship with Uniola, and they were sisters to Enneapogon. E. caerulescens, E. desvauxii, and E. oblongus formed a monophyletic cluster. In Eragrostis, E. setifolia diverged first. It showed a monophyletic relationship with the other Eragrostis species. E. mionor and E. autumnalis formed a monophyletic cluster, which was a sister to the other five Eragrostis species (E. tef, E. pilosa, E. cilianensis, E. nigra, and E. ferruginea). The monophyletic cluster comprising E. japonica and E. tenellula was a sister to the cluster composed of H. harpachnoides and E. tenella, and both were sisters to the other four Eragrostis species (E. unioloides, E. brownie, E. atrovirens, and E. fractus). H. harpachnoides was embedded in Eragrostis with high bootstrap values in all ML analyses (BS > 95%; Figure 8 and Figures S2–S7). In addition, we anatomized the morphology of H. harpachnoides and compared it with its sister group E. tenella in the phylogenetic tree we generated (Figure 1). In the early taxonomic period, Harpachne was recognized as a separate genus due to its racemes being completely different from the panicles of Eragrostis (Figure 1A). However, for E. japonica, E. tenellula, and E. tenella, florets disarticulated from above, moved downward, and fell together with the rachilla joints, and an analogous character—that spikelets fall entire together with pedicel—is found in H. harpachnoides (Figure 1B). Furthermore, Harpachne has characteristics including a ciliated ligule and three-veined lemmas (Figure 1C), which are very similar to other Eragrostis species.
3. Discussion
3.1. Basic Information on the Chloroplast Genomes of Eragrostideae
Chloroplast genomes have the characteristics of small size and a highly conserved structure [4,5]. In this study, chloroplast genomes were conservative in genome size, gene number, and GC content among 20 Eragrostideae species (Table 2), which was consistent with previous Eragrostideae plastome studies [3]. The chloroplast genomes of these species are approximately 134 kb in genome size (Figure S1). The GC content in each species was ca. 38%. Compared with other regions, the IR region had the highest GC content, possibly due to the fact may be because all rRNAs are located in this region [26]. In terms of the gene numbers, protein-coding genes had a small difference (83–87). The number of rRNA and tRNA genes were eight and 38, respectively. Due to the presence of mutations, insertions, and deletions, rps15 was not annotated in E. tenellula. A previous study reported the loss of accD, ycf1, and ycf2 genes in the family Poaceae [7]. The gene accD might be a useful molecular marker for phylogenetic analysis of land plants and is essential for leaf development [27]. In our study, we found that the accD gene had completely degraded in all species. Furthermore, in the case of ycf1 and ycf2 loss, there has been a progressive degradation of the gene sequences because different lengths of ycf1 and ycf2 were found in our study. The ycf1 gene has only been annotated in seven species, and retained segments of different lengths range from 78 to 135 bp. The ycf2 gene has retained segments of different lengths ranging from 105 to 477 bp. Both ycf1 and ycf2 have been reported to be essential genes in plants, but their functions are unclear [28,29]. Intron loss of clpP and rpoC1 was detected in all 20 Eragrostideae species. Gene and intron losses can lead to a decrease in chloroplast genome size.
Repeat sequences are not only hotspots where mutations such as nucleotide substitutions and insertion deletions occur, but also very important in phylogenetic research [30]. Repeat sequences are one of the most effective methods to study the origin and evolution of species at the molecular level [31,32]. In this study, 933 repeats were identified in 20 Eragrostideae chloroplast genomes and divided into four types: forward, palindromic, reverse, and complementary. Most of the repeats were forward and reverse repeats. Like most chloroplast genomes of angiosperms [33], most of repeat sequences in the chloroplast genome of Eragrostideae were located in noncoding regions. All the identified repeats in this study may be useful in population genetics studies of these 20 species in the future. SSRs are tandemly repeated DNA sequences, which are widely distributed in the genomes of eukaryotes. The SSRs in the plant chloroplast genomes are rich in genetic variation and have been widely proven as a high-resolution tool for revealing chloroplast genome variation [8]. We detected 943 SSRs in 20 Eragrostideae chloroplast genomes. Similar to the previously reported chloroplast genomes of angiosperms [8,34,35], the predominant type of SSRs were mono-nucleotides, of which, A or T repeats account for the majority. The SSRs detected in the Eragrostideae chloroplast genomes were of great significance for the phylogenetic research and classification of Eragrostideae plants.
Codon usage bias (CUB) is widespread in animals, plants, bacteria, and fungi, reflecting different pressures on different genes or genomes in the evolutionary process. CUB is specific among different species and even between different genes within a species, which is the result of the combined effects of mutation, selection, and drift in the long-term evolution of genes and species [36,37,38]. The results of this study indicated that the CUB of the Eragrostideae chloroplast genomes was weak. Base composition is one of the most pervasive effects of codon usage. In this study, GC content was highly conserved. It is consistent with previously reported Poaceae plastomes that all the 20 plastomes had similar codon usage patterns and preferred to use A/T-terminated codons (Table 4 and Table S1) [39,40]. In all species, nearly all the amino acid codons had a bias (RSCU > 1 or RSCU < 1), except for methionine (AUG, RSCU = 1) and tryptophan (UGG, RSCU = 1). This study can lay the foundation for further research and application of chloroplast genome codons in Eragrostideae.
In previous studies [41,42], the phylogenetic evolution of the chloroplast genome structure in Poaceae plants was reported, and it was found that the LSC/IR boundary had expanded and caused rps19 and trnH to move to the IR region. In addition, the border of SSC/IRa in the ancestors of Poaceae had expanded, resulting in rps15 being located in the IR region. On the PACMAD (Panicoideae, Aristidoideae, Chloridoideae, Micrairoideae, Arundinoideae, and Danthonioideae) clade of Poaceae, a part of the ndhF gene was duplicated at the IRb/SSC border, resulting in the border being located inside the ndhF gene [43]. In our present study, this phenomenon was observed in most species, with the exception of En. oblongus (Figure 5). The differences in the chloroplast genome length (130,773–135,322 bp) of different Eragrostideae plants were mainly caused by the expansion and contraction of the IR region. In general, the contraction and expansion of the IR regions are relatively common evolutionary events in plants and may play an important role in the evolution of plants [44,45].
3.2. Phylogenetically Informative Markers
With the continuous deepening study of plant chloroplast genomes, comparative analysis of chloroplast genomes has been paid more and more attention by researchers. Compared with other molecular markers, SNPs have the characteristics of high resolution and genetic stability. In addition, SNPs are unevenly distributed, and most of them are located in noncoding regions [9,46]. In this study, comparative analysis of complete chloroplast genomes in 20 Eragrostideae species showed that IR regions were more conservative than SC regions (Figure 6), and comparisons of the percentage of parsimony information sites confirmed that noncoding regions had higher Pi% than protein-coding genes. A total of 10 regions (rpl22, rpoA, ndhF, matK, trnG-UCC-trnT-GGU, ndhF-rpl32, ycf4-cemA, rpl32-trnL-UAG, trnG-GCC-trnfM-CAU, and ccsA-ndhD) with high Pi% were detected, of which, four regions (rpl22, rpoA, ndhF, and matK) were located in the coding regions and the rest (trnG-UCC-trnT-GGU, ndhF-rpl32, ycf4-cemA, rpl32-trnL-UAG, trnG-GCC-trnfM-CAU, and ccsA-ndhD) were located in the noncoding regions (Figure 7). Among the 10 potential phylogenetic informative markers, the region ndhF-rpl32 has been reported as a highly variable marker to study phylogenetic relationships among Eragrostideae species [3]. Understanding and using these variation hotspots is not only helpful for understanding the evolutionary characteristics of the Eragrostideae chloroplast genomes, but also can design molecular markers to provide a data basis for the classification and phylogeny of Eragrostideae.
3.3. Phylogenetic Relationships of Eragrostideae
In this study, Eragrostideae were divided into three clades (Eragrostis-Harpachne clade, Uniola clade, and Enneapogon clade), representing the three subtribes (Eragrostidinae, Uniolinae, Cotteinae). This was consistent with most previous studies on the phylogeny of Eragrostideae [12,13,14]. Eragrostis is the most widely distributed genus with the largest number of species in Eragrostideae, and the interspecies phylogenetic relationships are complicated. Many scholars advocated that several small genera such as Acamptoclados, Cladoraphis, Diandrochloa, Ectrosia, Psammagrostis, Harpachne, and Neeragrostis should be reclassified into Eragrostis [21,23,47]. In our study, we can intuitively see from all the ML trees that H. harpachnoides was sister to E. tenella with high support (BS > 95%; Figure 8 and Figures S2–S7). H. harpachnoides was embedded within Eragrostis, which was consistent with a previous study using the ITS and plastid sequences [12]. Morphologically, the raceme is a simplified structure of the panicle. In addition, H. harpachnoides is similar or even identical to the species of Eragrostis in the characteristics of spikelet drop patterns, ligule, and lemma. Based on the morphological and molecular evidence from this study, we suggest the reclassification of Eragrostis, including Harpachne. Few studies have been conducted on the phylogenetic relationship among Eragrostis species [3,21,23]. In our research, similar topologies were observed in the phylogenetic analysis of Eragrostideae based on different datasets of complete chloroplast genomes. Our study found that E. tef was a sister to E. polisa, and E. minor was a sister to E. autumnalis, which was consistent with a previous study [3]. In addition, our phylogenetic tree supported the clade comprising H. harpachnoides and E. tenella to be the sister to the clade composed of E. japonica and E. tenellula. The same result was obtained in previous studies by using nuclear and chloroplast gene data [24,48]. The interspecies relationships of Eragrostis were well resolved based on complete chloroplast genomes. This study indicated that complete chloroplast genomes could be used as super-barcodes to resolve the intergeneric and interspecies relationships within Eragrostideae. Moreover, broad sampling and more evidence from morphology and genomes will be necessary for further study of the interspecies relationships within Eragrostideae.
4. Materials and Methods
4.1. Plant Material, DNA Extraction, and Sequencing
Chloroplast genomes of 11 Eragrostis species, Enneapogon desvauxii, and Harpachne harpachnoides were newly sequenced. Plant material used in this study was deposited at the herbarium of the College of Life Sciences, Shandong Normal University. The voucher specimen information is presented in Table 1. Total genomic DNA was extracted by the modified CTAB method [49]. The total genomic DNA was used for library preparation and paired-end (PE) sequencing by the Illumina Novaseq instrument at Novogene (Beijing, China). Plastomes of other 11 species were downloaded from GenBank (E. minor (NC_029412), E. setifolia (NC_042832), E. tef (NC_029413), E. tenellula (NC_042833), E. caerulescens (NC_042837), E. oblongus (NC_036682), U. paniculata (NC_036709), Neyraudia reynaudiana (NC_024262), Triraphis mollis (NC_042835), Centropodia glauca (NC_029411), and Danthonia californica (NC_025232)).
4.2. Genome Assembly and Annotation
We assembled the chloroplast genome through Organelle Genome Assembler (OGA) [50], as described in Qu et al. [51]. Annotation was performed by using Plastid Genome Annotator (PGA) [52]. Geneious v8.0.2 was used for manual annotation correction [53]. The circular maps for newly sequenced plastomes were generated using the OGDRAW tool [54].
4.3. Genome Structure and Expansion and Contraction of IR Region
The chloroplast genome structure, such as gene length, gene number, GC content, intron number were summarized and comparatively analyzed by Geneious v8.0.2. The expansion and contraction of IR regions were analyzed by IRscope [55], coupled with manual modification.
4.4. Repeat Sequences and SSR Analysis
The size and position of the repeat sequences were detected using REPuter [56] with the following parameters: hamming distance of 3 and minimum repeat size of 30 bp [56]. MISA script [57] was used to detect SSR, and the minimum number of repeats were set as 10, 6, 5, 5, 5, and 5 for mono-, di-, tri-, tetra-, penta-, and hexanucleotide SSRs, respectively.
4.5. Codon Usage
Codons encoding the same amino acid are called synonymous codons, and the difference in use frequency of synonymous codons is the CUB. In order to ensure the accuracy of the results, we eliminated sequences less than 300 bp before codon analysis [58,59]. Then, the codon usage frequency was calculated using codonW v1.4.2 [60]. We also analyzed the effective number of codons (ENC) [61] and relative synonymous codon usage (RSCU). ENC refers to the effective number of codons, and the range of its theoretical value is 20–61, representing the strength of codon bias. The larger the value, the weaker the codon bias. RSCU refers to the relative probability between synonymous codons encoding corresponding amino acids for a particular codon. If there is no preference for the use of codons, the RSCU value of the codon is equal to 1.00. When the RSCU value of a codon is greater than 1.00, it indicates that the frequency of the codon use is relatively high, and vice versa.
4.6. Comparative Genome Analysis and Divergent Hotspot Regions
mVISTA [62] is a commonly used comparative chloroplast genome map-drawing program, but the input file needs to meet the format requirements of mVISTA. For comparative analysis, a script [63] was used to convert GenBank annotation files to mVISTA format files. Then, we aligned the complete chloroplast genomes using the mVISTA program with the Shuffle-LAGAN mode, with E. atrovirens as a reference [62].
Single nucleotide polymorphism (SNP) mainly refers to a DNA sequence polymorphism caused by single nucleotide variation at the genome level. We counted and calculated the percentage of parsimony information sites (Pi%) of the selected CDS and noncoding regions by using MEGA v7.0.26 [64]. The screening conditions were as follows: (a) sequence length > 200 bp; (b) variable sites and parsimony information sites > 0 [65].
4.7. Phylogenetic Analysis
In this study, we downloaded the complete chloroplast genomes of seven Eragrostideae species (Chloridoideae), one Centropodieae species (Chloridoideae), two Triraphideae species (Chloridoideae), and one Danthonieae species (Danthonioideae) from GenBank. D. californica was set as outgroup. Sequences were aligned using MAFFT v7.313 [66] with default parameters. The ML trees [1] were reconstructed by RAxML v8.0.26 with the GTRGAMMA substitution model and 1000 bootstrap replicates [67]. Seven trees were reconstructed based on following seven data sets: (1) complete chloroplast genome sequences; (2) coding sequences; (3) noncoding sequences; (4) hypervariable regions (rpl22, rpoA, ndhF, matK, trnG-UCC-trnT-GGU, ndhF-rpl32, ycf4-cemA, rpl32-trnL-UAG, trnG-GCC-trnfM-CAU, and ccsA-ndhD); (5) LSC regions; (6) SSC regions; (7) IR regions.
5. Conclusions
In this study, 13 Eragrostideae chloroplast genomes were assembled. Combining the downloaded sequences, a total of 20 Eragrostideae chloroplast genomes were collected. All the plastomes were conserved in structure, gene content, gene order, and IR boundaries. Repeats and SSRs were identified, which are important to study chloroplast genome evolution. By examining parsimony information sites, 10 highly variable regions were identified, which can be used as candidate molecular markers for phylogenetic and population genetics study of Eragrostideae. Our phylogenetic analysis found that three species of Enneapogon formed a monophyletic clade. Enneapogon diverged first, and Eragrostis, including Harpachne, is the sister to Uniola. In addition, the interspecies relationships of Eragrostis were well resolved. E. setifolia was suggested to be an early-diverging species. Furthermore, H. harpachnoides was considered as a species of Eragrostis based on morphological and molecular evidence. This is the first time that the complete chloroplast genomes support the clustering of Harpachne into Eragrostis. Our results presented here, provide helpful insights into the phylogenetic study of Eragrostideae species based on complete chloroplast genomes. Moreover, broad sampling and more evidence will be necessary for further study into the relationships of Eragrostideae.
Supplementary Materials
The following are available online at https://www.mdpi.com/2223-7747/10/1/109/s1, Figure S1: Chloroplast genome maps of 13 Eragrostideae species. Genes on the inside of outer circle are transcribed in the clockwise direction and genes on the outside are transcribed in the counterclockwise direction. The dashed darker gray area in the inner circle indicates GC content, while the lighter gray area shows AT content. IR = inverted repeat; SSC = small single copy; LSC = large single copy. Figure S2: The ML tree of 20 Eragrostideae species based on the coding sequences. The numbers next to the branches are bootstrap support values. Figure S3: The ML tree of 20 Eragrostideae species based on the noncoding sequences. The numbers next to the branches are bootstrap support values. Figure S4: The ML tree of 20 Eragrostideae species based on the hypervariable regions (rpl22, rpoA, ndhF, matK, trnG-UCC-trnT-GGU, ndhF-rpl32, ycf4-cemA, rpl32-trnL-UAG, trnG-GCC-trnfM-CAU, and ccsA-ndhD). The numbers next to the branches are bootstrap support values. Figure S5: The ML tree of 20 Eragrostideae species based on the LSC regions. The numbers next to the branches are bootstrap support values. Figure S6: The ML tree of 20 Eragrostideae species based on the SSC regions. The numbers next to the branches are bootstrap support values. Figure S7: The ML tree of 20 Eragrostideae species based on the IR regions. The numbers next to the branches are bootstrap support values. Table S1: types of codon encoding amino acids, Table S2: codon usage condition in 20 Eragrostideae chloroplast genomes.
Author Contributions
Conceiving and designing, X.-J.Q., and S.-J.F.; performing and analyzing data, K.L.; writing—original draft preparation, K.L.; writing—review and editing, K.L., R.W., and X.-X.G.; supervision, X.-J.Q., X.-J.Z., and S.-J.F.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China, grant number 31470298 and Shandong Agricultural Science and Technology Fund Project, grant number 2019LY002.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are openly available in GeneBank.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; dePamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar]
- Shaw, J.; Lickey, E.; Schilling, E.; Small, R. Comparison of whole chloroplast genome sequence to choose noncoding regions for phylogenetic studies in angiosperms. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [Green Version]
- Somaratne, Y.; Guan, D.-L.; Abbood, N.N.; Zhao, L.; Xu, S.-Q. Comparison of the complete Eragrostis pilosa chloroplast genome with its relatives in Eragrostideae (Chloridoideae; Poaceae). Plants 2019, 8, 485. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wang, Q.-J.; Qu, X.-J.; Fan, S.-J. Characterization of the complete plastome of Alopecurus aequalis (Poaceae), a widespread weed. Mitochondrial DNA Part B 2019, 4, 4216–4217. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.-X.; Dai, C.; Wang, R.; Qu, X.-J.; Zhang, X.-J. Characterization and phylogenetic analysis of the complete plastome of Alopecurus japonicus (Gramineae), an annual weed. Mitochondrial DNA Part B 2020, 5, 396–397. [Google Scholar] [CrossRef] [Green Version]
- Leseberg, C.H.; Duvall, M.R. The complete chloroplast genome of Coix lacryma-jobi and a comparative molecular evolutionary analysis of plastomes in cereals. J. Mol. Evol. 2009, 69, 311–318. [Google Scholar] [CrossRef]
- Tang, P.; Ruan, Q.; Peng, C. Phylogeny in structure alterations of Poaceae cpDNA. Chin. Agric. Ence Bull. 2011, 27, 171–176. [Google Scholar]
- Chakravarthi, B.K.; Naravaneni, R. SSR marker based DNA fingerprinting and diversity study in rice (Oryza sativa. L). Afr. J. Biotechnol. 2006, 5, 684–688. [Google Scholar]
- Cesare, M.D.; Hodkinson, T.; Barth, S. Chloroplast DNA markers (cpSSRs, SNPs) for Miscanthus, Saccharum and related grasses (Panicoideae, Poaceae). Mol. Breed. 2010, 26, 539–544. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, N. Change and development of the classification systems of the subfamily Chloridoideae (Gramineae). J. Trop. Subtrop. Bot. 2004, 12, 91–98. [Google Scholar]
- Liu, Q.; Zhao, N.; Hao, G. The phylogeny of the Chloridoideae (Gramineae): A cladistic analysis. J. Trop. Subtrop. Bot. 2005, 13, 432–442. [Google Scholar]
- Peterson, P.M.; Romaschenko, K.; Johnson, G. A classification of the Chloridoideae (Poaceae) based on multi-gene phylogenetic trees. Mol. Phylogenetics Evol. 2010, 55, 580–598. [Google Scholar] [CrossRef]
- Soreng, R.J.; Peterson, P.M.; Romaschenko, K.; Davidse, G.; Zuloaga, F.O.; Judziewicz, E.J.; Filgueiras, T.S. A worldwide phylogenetic classification of the Poaceae (Gramineae). J. Syst. Evol. 2015, 53, 117–137. [Google Scholar] [CrossRef]
- Soreng, R.J.; Peterson, P.M.; Romaschenko, K.; Davidse, G.; Teisher, J.K.; Clark, L.G.; Barberá, P.; Gillespie, L.J.; Zuloaga, F.O. A worldwide phylogenetic classification of the Poaceae (Gramineae) II: An update and a comparison of two 2015 classifications. J. Syst. Evol. 2017, 55, 259–290. [Google Scholar] [CrossRef] [Green Version]
- Ellis, R.P. Eragrostis walteri—A first record of non-Kranz leaf anatomy in the sub-family Chloridoideae (Poaceae). S. Afr. J. Bot. 1984, 3, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Ingram, A.L.; Christin, P.-A.; Osborne, C.P. Molecular phylogenies disprove a hypothesized C4 reversion in Eragrostis walteri (Poaceae). Ann. Bot. 2010, 107, 321–325. [Google Scholar] [CrossRef]
- Chen, S.; Li, D.; Zhu, G.; Wu, Z.; Lu, S.; Liu, L.; Wang, C.; Sun, B.; Chu, C.; Xia, N.; et al. POACEAE (GRAMINEAE). In Flora of China; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MI, USA, 2006; pp. 1–653. [Google Scholar]
- Kellogg, E.A.X. Subfamily Chloridoideae Kunth ex Beilschm. (1833). In Flowering Plants Monocots; Springer International Publishing: Berlin, Germany, 2015; pp. 353–397. [Google Scholar]
- Sutherland, D.M. Genera Graminum. Grasses of the World. Brittonia 1986, 39, 508. [Google Scholar] [CrossRef]
- Peterson, P. Catalogue of new world grasses (Poaceae): II. subfamily Chloridoideae. Contrib. U. S. Natl. Herb. 2001, 41, 176–177. [Google Scholar]
- Ingram, A.L.; Doyle, J.J. Eragrostis (Poaceae): Monophyly and infrageneric classification. Aliso A J. Syst. Evol. Bot. 2007, 23, 595–604. [Google Scholar] [CrossRef] [Green Version]
- Ingram, A.L.; Doyle, J.J. The origin and evolution of Eragrostis tef (Poaceae) and related polyploids: Evidence from nuclear waxy and plastid rps16. Am. J. Bot. 2003, 90, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Ingram, A.L.; Doyle, J.J. Is Eragrostis (Poaceae) monophyletic? Insights from nuclear and plastid sequence data. Syst. Bot. 2004, 29, 545–552. [Google Scholar] [CrossRef]
- Barrett, R.L.; Peterson, P.M.; Romaschenko, K. A molecular phylogeny of Eragrostis (Poaceae: Chloridoideae: Eragrostideae): Making lovegrass monophyletic in Australia. Aust. Syst. Bot. 2020, 33, 458–476. [Google Scholar] [CrossRef]
- Columbus, J.T.; Cerros-Tlatilpa, R.; Kinney, M.S.; Siqueiros-Delgado, M.E.; Bell, H.L.; Griffith, M.P.; Refulio-Rodriguez, N.F. Phylogenetics of Chloridoideae (Gramineae): A preliminary study based on nuclear ribosomal internal transcribed spacer and chloroplast trnL–F sequences. Aliso A J. Syst. Evol. Bot. 2007, 23, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Penaflor, C.; Kuehl, J.V.; Leebens-Mack, J.; Carlson, J.E.; dePamphilis, C.W.; Boore, J.L.; Jansen, R.K. Complete plastid genome sequences of Drimys, Liriodendron, and Piper: Implications for the phylogenetic relationships of magnoliids. BMC Evol. Biol. 2006, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef]
- Kikuchi, S.; Bédard, J.; Hirano, M.; Hirabayashi, Y.; Oishi, M.; Imai, M.; Takase, M.; Ide, T.; Nakai, M. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 2013, 339, 571–574. [Google Scholar] [CrossRef]
- Drescher, A.; Ruf, S.; Calsa, T.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. Cell Mol. Biol. 2000, 22, 97–104. [Google Scholar] [CrossRef]
- Asaf, S.; Waqas, M.; Khan, A.L.; Khan, M.A.; Kang, S.-M.; Imran, Q.M.; Shahzad, R.; Bilal, S.; Yun, B.-W.; Lee, I.-J. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front. Plant Sci. 2017, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Cavalier-Smith, T. Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 2002, 12, 62–64. [Google Scholar] [CrossRef] [Green Version]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [Green Version]
- Vu, H.T.; Tran, N.; Nguyen, T.D.; Vu, Q.L.; Bui, M.H.; Le, M.T.; Le, L. Complete chloroplast genome of Paphiopedilum delenatii and phylogenetic relationships among Orchidaceae. Plants 2020, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provan, J.; Soranzo, N.; Wilson, N.; Goldstein, D.; Powell, W. A low mutation rate for chloroplast microsatellites. Genetics 1999, 153, 943–947. [Google Scholar]
- Provan, J.; Powell, W.; Hollingsworth, P. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Morton, B. The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J. Mol. Evol. 2003, 56, 616–629. [Google Scholar] [CrossRef]
- Netherlands, S. Codon usage. In Encyclopedia of Genetics, Genomics, Proteomics and Informatics; Springer: Dordrecht, The Netherlands, 2008; pp. 383–384. [Google Scholar]
- Shang, M.Z.; Liu, F.; Hua, J.P.; Wang, K.B. Analysis on codon usage of chloroplast genome of Gossypium hirsutum. Sci. Agric. Sin. 2011, 44, 245–253. [Google Scholar]
- Sajjad, A.; Khan, A.L.; Khan, A.R.; Muhammad, W.; Kang, S.M.; Khan, M.A.; Seok-Min, L.; In-Jung, L. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 2016, 7, 843. [Google Scholar]
- Yin, D.; Wang, Y.; Zhang, X.; Ma, X.; He, X.; Zhang, J. Development of chloroplast genome resources for peanut (Arachis hypogaea L.) and other species of Arachis. Sci. Rep. 2017, 7, 11649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, H.; Ogihara, Y. Structural alterations of the chloroplast genome found in grasses are not common in monocots. Curr. Genet. 1993, 23, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Ogihara, Y. Phylogenetic affinities of the grasses to other monocots as revealed by molecular analysis of chloroplast DNA. Curr. Genet. 1996, 29, 572–581. [Google Scholar] [CrossRef]
- Davis, J.; Soreng, R. Migration of endpoints of two genes relative to boundaries between regions of the plastid genome in the grass family (Poaceae). Am. J. Bot. 2010, 97, 874–892. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Ma, P.-F.; Wen, J.; Yi, T.-S. Complete sequencing of five Araliaceae chloroplast genomes and the phylogenetic implications. PLoS ONE 2013, 8, e78568. [Google Scholar] [CrossRef]
- Zheng, X.M.; Wang, J.; Li, F.; Sha, L.; Pang, H.; Lan, Q.; Jing, L.; Yan, S.; Qiao, W.; Zhang, L. Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 2017, 7, 1555. [Google Scholar]
- Rajicic, T.; Lübberstedt, T.; Jensen, L.; Scholz, U.; Weber, W.; Graner, A.; Dehmer, K. Single nucleotide polymorphism (SNP) markers for allele quantification in Lolium (Poaceae): Development and first applications. In Molecular Breeding of Forage and Turf; Springer: Berlin, Germany, 2015; pp. 143–163. [Google Scholar]
- Fisher, A.E.; Hasenstab, K.M.; Bell, H.L.; Blaine, E.; Ingram, A.L.; Columbus, J.T. Evolutionary history of chloridoid grasses estimated from 122 nuclear loci. Mol. Phylogenet. Evol. 2016, 105, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, R. Systematic Studies on Eragrostis; Shandong Normal University: Jinan, China, 2017. [Google Scholar]
- Doyle, J.; Doyle, J. A rapid DNA isolation procedure from small quantities of fresh leaf tissues. Polym. Bull. 1987, 19, 11–15. [Google Scholar]
- Qu, X.-J. Organelle Genome Assembler. Available online: https://github.com/quxiaojian/OGA (accessed on 24 May 2020).
- Qu, X.-J.; Fan, S.-J.; Wicke, S.; Yi, T.-S. Plastome reduction in the only parasitic gymnosperm Parasitaxus is due to losses of photosynthesis but not housekeeping genes and apparently involves the secondary gain of a large inverted repeat. Genome Biol. Evol. 2019, 11, 2789–2796. [Google Scholar] [CrossRef]
- Qu, X.-J.; Moore, M.; Li, D.-Z.; Yi, T. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Frank, W. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar]
- Rosenberg, M.; Subramanian, S.; Kumar, S. Patterns of transitional biases within and among mammalian genomes. Mol. Biol. Evol. 2003, 20, 988–993. [Google Scholar] [CrossRef]
- Peden, J. CodonW. Available online: http://codonw.sourceforge.net/index.html (accessed on 27 May 2020).
- Hilu, K.W.; Alice, L.A.; Liang, H. Phylogeny of Poaceae inferred from matK sequences. Ann. Mo. Bot. Gard. 1999, 86, 835–851. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Qu, X.-J. Get mVISTA Format from GenBank Annotation. Available online: https://github.com/quxiaojian/Bioinformatic_Scripts/tree/master/get_mVISTA_format_from_GenBank_annotation (accessed on 10 June 2020).
- Bingjuan, L.; Guohui, X.; Zaien, X.; Xiaoqin, G. Universal genetic markers for the Poaceae family. J. Zhejiang A&F Univ. 2014, 31, 508–514. [Google Scholar]
- Chen, C.; Zheng, Y.; Liu, S.; Zhong, Y.; Wu, Y.; Li, J.; Xu, L.-A.; Xu, M. The complete chloroplast genome of Cinnamomum camphora and its comparison with related Lauraceae species. PeerJ 2017, 5, e3820. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
Figure 1.
The flower structures of Harpachne harpachnoides (left) and Eragrostis tenella (right). (A) Inflorescence structure; (B) Spikelets structure; (C) Floret structure.
Figure 1.
The flower structures of Harpachne harpachnoides (left) and Eragrostis tenella (right). (A) Inflorescence structure; (B) Spikelets structure; (C) Floret structure.

Figure 2.
The number of four repeat types in 20 Eragrostideae chloroplast genomes.

Figure 3.
The number of repeats with different length in 20 Eragrostideae chloroplast genomes.

Figure 4.
Amino acid proportion of protein-coding genes in 20 Eragrostideae chloroplast genomes.

Figure 5.
Comparison of the junctions of LSC, SSC, and IR regions among 20 Eragrostideae chloroplast genomes.
Figure 5.
Comparison of the junctions of LSC, SSC, and IR regions among 20 Eragrostideae chloroplast genomes.

Figure 6.
Comparison of 20 Eragrostideae chloroplast genomes using mVISTA alignment program with E. atrovirens as a reference. Genome regions are color-coded: blue blocks, exons of protein-coding genes (exon); sky-blue blocks, tRNA and rRNA genes; red blocks, conserved noncoding sequences (CNS). The vertical scale represents a 50–100% of identity.
Figure 6.
Comparison of 20 Eragrostideae chloroplast genomes using mVISTA alignment program with E. atrovirens as a reference. Genome regions are color-coded: blue blocks, exons of protein-coding genes (exon); sky-blue blocks, tRNA and rRNA genes; red blocks, conserved noncoding sequences (CNS). The vertical scale represents a 50–100% of identity.

Figure 7.
Comparison of percentage of parsimony information sites (Pi%) in 20 Eragrostideae chloroplast genomes. (A) Pi% values among coding sequences (CDS); (B) Pi% values among noncoding regions (IGS and intron regions).
Figure 7.
Comparison of percentage of parsimony information sites (Pi%) in 20 Eragrostideae chloroplast genomes. (A) Pi% values among coding sequences (CDS); (B) Pi% values among noncoding regions (IGS and intron regions).

Figure 8.
The maximum likelihood phylogenetic tree of 20 Eragrostideae species based on the complete chloroplast genomes. The numbers next to the branches are bootstrap support (BS) values.
Figure 8.
The maximum likelihood phylogenetic tree of 20 Eragrostideae species based on the complete chloroplast genomes. The numbers next to the branches are bootstrap support (BS) values.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Voucher specimen information of newly sequenced 13 Eragrostideae species.
| Species | Collecting Locations | GPS | Voucher Specimen Number | GenBank Accession Number |
|---|---|---|---|---|
| Eragrostis atrovirens | Guangdong, China | 113°46″ E, 23°29′ N | SDNU101 | MW255512 |
| E. autumnalis | Gansu, China | 105°53′ E, 34°34′ N | SDNU012 | MW255513 |
| E. brownii | Guangdong, China | 111°57′ E, 22°42′ N | SDNU022 | MW255514 |
| E. cilianensis | Shandong, China | 118°36′ E, 36°12′ N | SDNU235 | MW255515 |
| E. ferruginea | Shandong, China | 117°20′ E, 36°29′ N | SDNU002 | MW255517 |
| E. fractus | Yunnan, China | 100°11′ E, 25°38′ N | SDNU184 | MW255518 |
| E. japonica | Guangdong, China | 111°57′ E, 22°42′ N | SDNU013 | MW255519 |
| E. nigra | Yunnan, China | 100°11′ E, 25°38′ N | SDNU183 | MW255521 |
| E. pilosa | Guangdong, China | 113°46″ E, 23°29′ N | SDNU087 | MW255523 |
| E. tenella | Guangdong, China | 111°57′ E, 22°42′ N | SDNU011 | MW255525 |
| E. unioloides | Guangdong, China | 113°46′ E, 23°29′ N | SDNU003 | MW255526 |
| Harpachne harpachnoides | Yunnan, China | 100°11′ E, 25°38′ N | SDNU088 | MW255527 |
| Enneapogon desvauxii | Inner Mongolia, China | 111°35′ E, 40°51′ N | SDNU046 | MW255511 |
Table 2.
Chloroplast genome characteristics of 20 Eragrostideae species.
| Species | Genome Size | LSC Region | IR Region | SSC Region | GC Content (%) | Number of Genes | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (bp) | (bp) | (bp) | (bp) | Overall | IR | LSC | SSC | Total | PCGs | rRNAs | tRNAs | |
| Eragrostis atrovirens | 134,857 | 80,113 | 21,038 | 12,668 | 38.2 | 44.0 | 36.1 | 32.0 | 133 | 87 | 8 | 38 |
| E. autumnalis | 134,556 | 79,861 | 21,025 | 12,645 | 38.3 | 44.0 | 36.2 | 32.1 | 131 | 85 | 8 | 38 |
| E. brownii | 134,728 | 80,098 | 21,016 | 12,598 | 38.2 | 44.0 | 36.1 | 32.1 | 131 | 85 | 8 | 38 |
| E. cilianensis | 134,654 | 80,005 | 21,026 | 12,597 | 38.2 | 44.0 | 36.2 | 32.1 | 131 | 85 | 8 | 38 |
| E. ferruginea | 134,380 | 79,732 | 21,028 | 12,592 | 38.2 | 44.0 | 36.1 | 32.2 | 131 | 85 | 8 | 38 |
| E. fractus | 134,578 | 79,894 | 21,058 | 12,568 | 38.2 | 43.9 | 36.1 | 32.0 | 133 | 87 | 8 | 38 |
| E. japonica | 134,126 | 79,323 | 21,074 | 12,655 | 38.2 | 43.9 | 36.2 | 32.1 | 133 | 87 | 8 | 38 |
| E. minor | 135,023 | 80,316 | 21,065 | 12,577 | 38.2 | 44.0 | 36.2 | 32.2 | 131 | 85 | 8 | 38 |
| E. nigra | 134,861 | 80,154 | 21,028 | 12,651 | 38.2 | 44.0 | 36.2 | 32.1 | 131 | 85 | 8 | 38 |
| E. pilosa | 134,737 | 80,098 | 21,026 | 12,587 | 38.2 | 44.0 | 36.2 | 32.0 | 131 | 85 | 8 | 38 |
| E. setifolia | 134,928 | 80,416 | 21,009 | 12,494 | 38.3 | 44.0 | 36.2 | 32.4 | 131 | 85 | 8 | 38 |
| E. tef | 134,435 | 79,802 | 21,026 | 12,581 | 38.3 | 44.0 | 36.3 | 32.1 | 131 | 85 | 8 | 38 |
| E. tenella | 134,550 | 79,876 | 21,027 | 12,620 | 38.2 | 43.9 | 36.2 | 32.2 | 133 | 87 | 8 | 38 |
| E. tenellula | 130,773 | 79,387 | 19,394 | 12,598 | 38.4 | 44.9 | 36.2 | 32.2 | 129 | 83 | 8 | 38 |
| E. unioloides | 134,711 | 80,088 | 21,016 | 12,591 | 38.2 | 44.0 | 36.1 | 32.2 | 131 | 85 | 8 | 38 |
| Harpachne harpachnoides | 133,830 | 79,083 | 21,069 | 12,609 | 38.2 | 43.9 | 36.2 | 32.3 | 133 | 87 | 8 | 38 |
| Enneapogoncaerulescens | 133,231 | 78,883 | 20,969 | 12,410 | 38.3 | 44.0 | 36.3 | 32.3 | 133 | 87 | 8 | 38 |
| E. desvauxii | 131,516 | 77,993 | 20,506 | 12,511 | 38.3 | 44.1 | 36.3 | 32.3 | 133 | 87 | 8 | 38 |
| E. oblongus | 133,433 | 78,839 | 21,024 | 12,546 | 38.3 | 44.0 | 36.3 | 32.3 | 133 | 87 | 8 | 38 |
| Uniola paniculata | 135,322 | 80,643 | 21,042 | 12,595 | 38.3 | 44.0 | 36.3 | 32.4 | 131 | 85 | 8 | 38 |
Table 3.
Numbers of SSRs in 20 Eragrostideae chloroplast genomes.
| Species | Total SSRs | Compound SSRs | A/T | C/G | AT/TA | LSC | SSC | IRa | IRb | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | T | C | G | AT | TA | |||||||
| Eragrostis atrovirens | 40 | 4 | 18 | 21 | - | - | - | 1 | 33 | 3 | 2 | 2 |
| E. autumnalis | 44 | 5 | 17 | 25 | 1 | 1 | - | - | 38 | 2 | 2 | 2 |
| E. brownii | 52 | 7 | 19 | 28 | 1 | 1 | 1 | 2 | 44 | 6 | 1 | 1 |
| E. cilianensis | 46 | 4 | 17 | 25 | 1 | 1 | - | 2 | 39 | 3 | 2 | 2 |
| E. ferruginea | 46 | 4 | 20 | 23 | 1 | 1 | - | 1 | 37 | 3 | 3 | 3 |
| E. fractus | 48 | 7 | 20 | 26 | - | 1 | - | 1 | 45 | 1 | 1 | 1 |
| E. japonica | 51 | 2 | 20 | 27 | 1 | 2 | - | 1 | 42 | 3 | 3 | 3 |
| E. minor | 41 | 5 | 15 | 23 | 1 | 1 | - | 1 | 36 | 1 | 2 | 2 |
| E. nigra | 53 | 5 | 20 | 29 | 2 | 2 | - | - | 43 | 4 | 3 | 3 |
| E. pilosa | 50 | 4 | 18 | 26 | 2 | 3 | - | 1 | 43 | 1 | 3 | 3 |
| E. setifolia | 40 | 3 | 15 | 23 | 1 | 1 | - | - | 33 | 2 | 2 | 2 |
| E. tef | 50 | 7 | 22 | 25 | 1 | 1 | - | 1 | 47 | 1 | 1 | 1 |
| E. tenella | 56 | 5 | 21 | 30 | 1 | 1 | 1 | 2 | 48 | 2 | 3 | 3 |
| E. tenellula | 49 | 2 | 18 | 27 | 1 | 2 | - | 1 | 42 | 3 | 2 | 2 |
| E. unioloides | 52 | 7 | 19 | 29 | - | 2 | - | 2 | 45 | 5 | 1 | 1 |
| Harpachne harpachnoides | 43 | 5 | 15 | 24 | 1 | 1 | - | 2 | 36 | 1 | 3 | 3 |
| Enneapogon caerulescens | 44 | 2 | 21 | 20 | - | - | - | 3 | 39 | 1 | 2 | 2 |
| E. desvauxii | 43 | 2 | 17 | 22 | 1 | 1 | - | 2 | 37 | - | 3 | 3 |
| E. oblongus | 45 | 2 | 21 | 21 | - | - | - | 3 | 40 | 1 | 2 | 2 |
| Uniola paniculata | 50 | 8 | 20 | 29 | - | - | - | 1 | 50 | - | - | - |
Table 4.
GC content, codons count, and effective number of codons (ENC) of protein-coding genes in 20 Eragrostideae chloroplast genomes.
Table 4.
GC content, codons count, and effective number of codons (ENC) of protein-coding genes in 20 Eragrostideae chloroplast genomes.
| Species | GC 1 | CC 2 | ENC 3 |
|---|---|---|---|
| Eragrostis atrovirens | 0.390 | 17,169 | 49.61 |
| E. autumnalis | 0.389 | 17,201 | 49.52 |
| E. brownii | 0.390 | 17,171 | 49.66 |
| E. cilianensis | 0.389 | 17,208 | 49.51 |
| E. ferruginea | 0.389 | 17,129 | 49.56 |
| E. fractus | 0.389 | 17,159 | 49.52 |
| E. japonica | 0.389 | 16,999 | 49.48 |
| E. minor | 0.390 | 17,160 | 49.61 |
| E. nigra | 0.389 | 17,208 | 49.59 |
| E. pilosa | 0.389 | 17,210 | 49.52 |
| E. setifolia | 0.391 | 17,149 | 49.73 |
| E. tef | 0.390 | 17,146 | 49.60 |
| E. tenella | 0.390 | 17,169 | 49.59 |
| E. tenellula | 0.390 | 17,123 | 49.52 |
| E. unioloides | 0.390 | 17,171 | 49.66 |
| Harpachne harpachnoides | 0.389 | 17,173 | 49.55 |
| Enneapogoncaerulescens | 0.390 | 17,158 | 49.40 |
| E. desvauxii | 0.390 | 17,145 | 49.42 |
| E. oblongus | 0.390 | 17,068 | 49.40 |
| Uniolapaniculata | 0.391 | 17,097 | 49.80 |
| Average | 0.3897 | 17,151 | 49.56 |
1 GC content at coding positions; 2 Codons count; 3 Effective number of codons.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, K.; Wang, R.; Guo, X.-X.; Zhang, X.-J.; Qu, X.-J.; Fan, S.-J. Comparative and Phylogenetic Analysis of Complete Chloroplast Genomes in Eragrostideae (Chloridoideae, Poaceae). Plants 2021, 10, 109. https://doi.org/10.3390/plants10010109
AMA Style
Liu K, Wang R, Guo X-X, Zhang X-J, Qu X-J, Fan S-J. Comparative and Phylogenetic Analysis of Complete Chloroplast Genomes in Eragrostideae (Chloridoideae, Poaceae). Plants. 2021; 10(1):109. https://doi.org/10.3390/plants10010109
Chicago/Turabian StyleLiu, Kuan, Rong Wang, Xiu-Xiu Guo, Xue-Jie Zhang, Xiao-Jian Qu, and Shou-Jin Fan. 2021. "Comparative and Phylogenetic Analysis of Complete Chloroplast Genomes in Eragrostideae (Chloridoideae, Poaceae)" Plants 10, no. 1: 109. https://doi.org/10.3390/plants10010109
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.