
Deciphering the Plastomic Code of Chinese Hog-Peanut (Amphicarpaea edgeworthii Benth., Leguminosae): Comparative Genomics and Evolutionary Insights within the Phaseoleae Tribe
 ,
,
Abstract
:1. Introduction
2. Results and Discussion
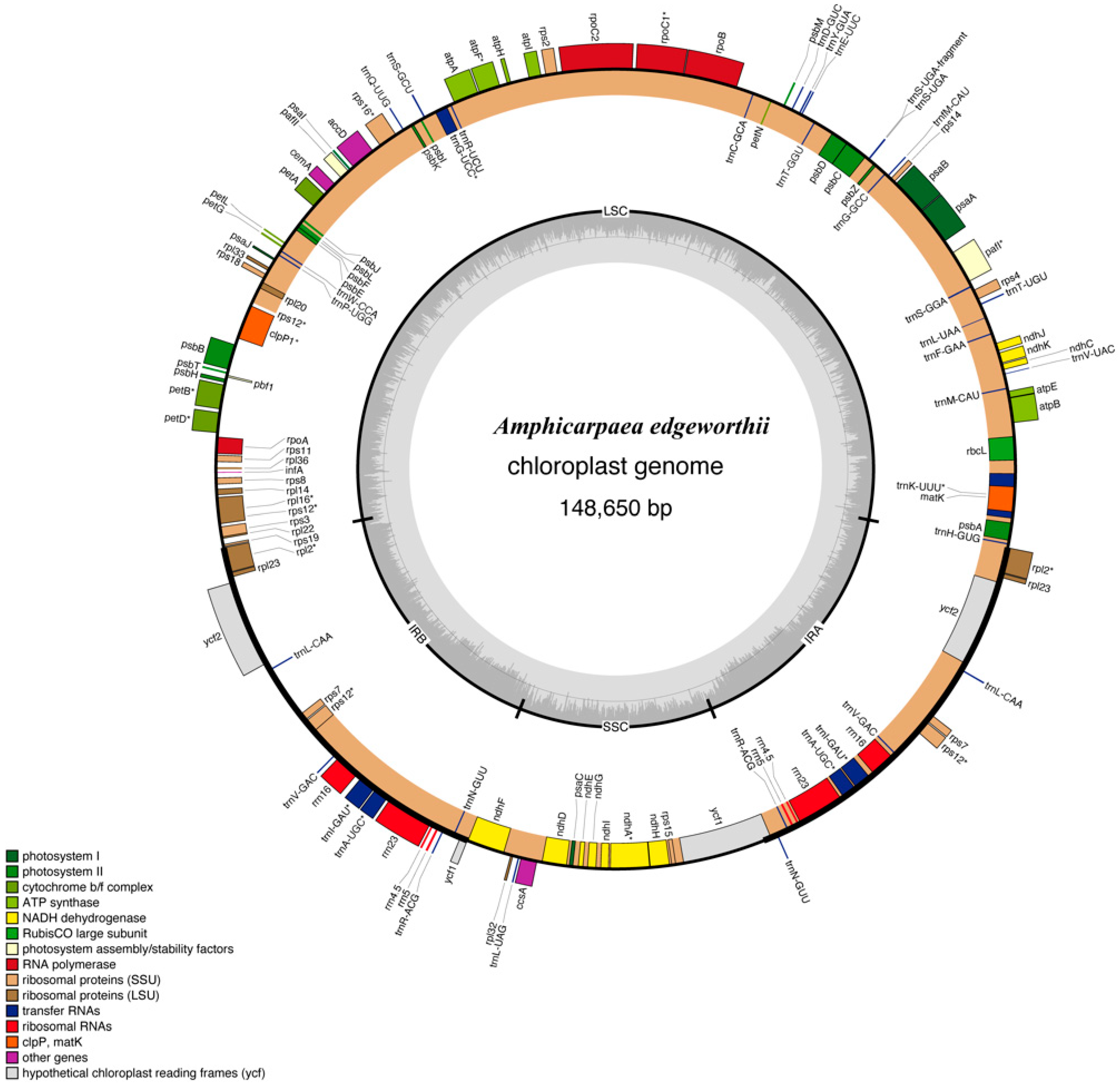
2.1. The Overall Structure and General Features of the A. edgeworthii Chloroplast Genome
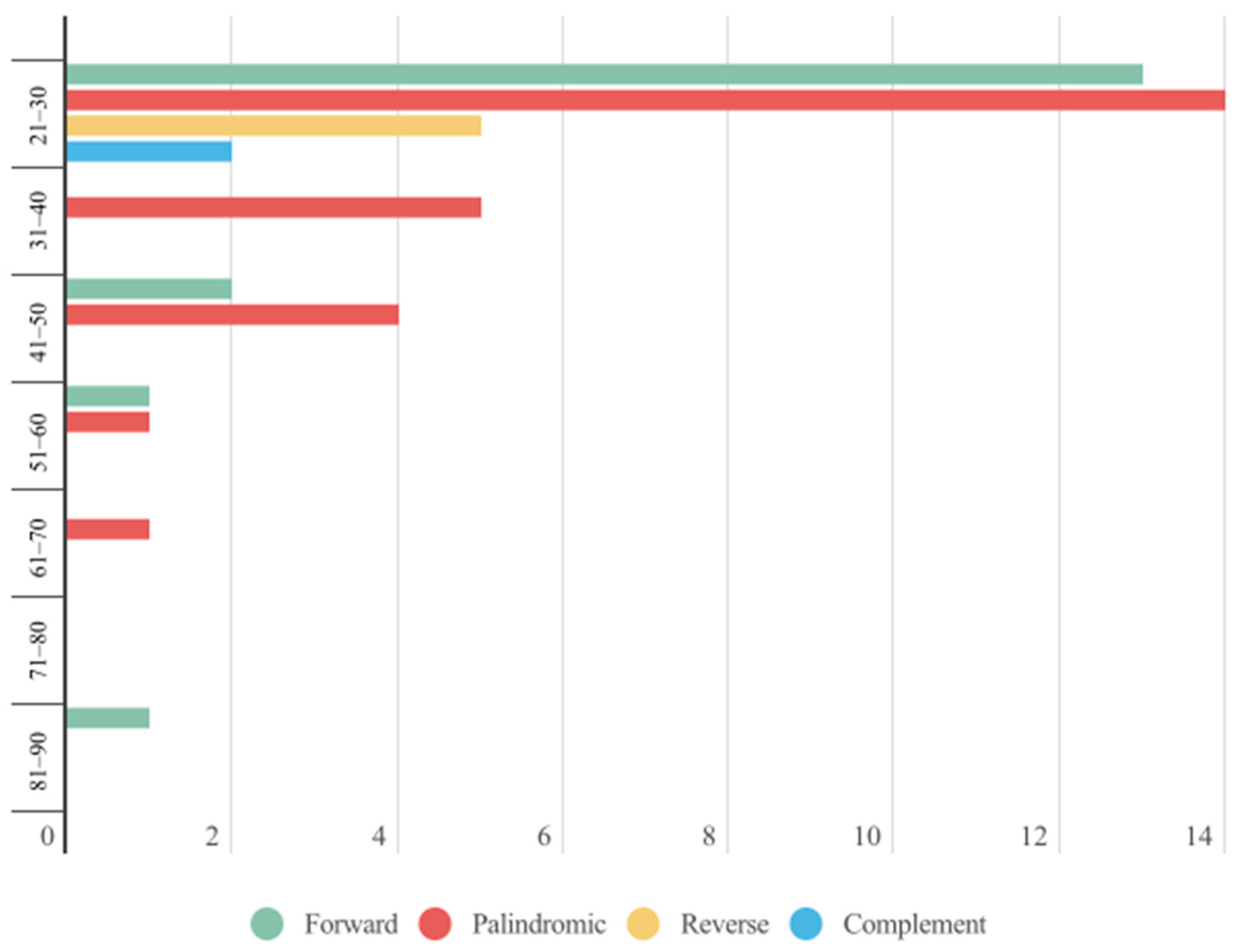
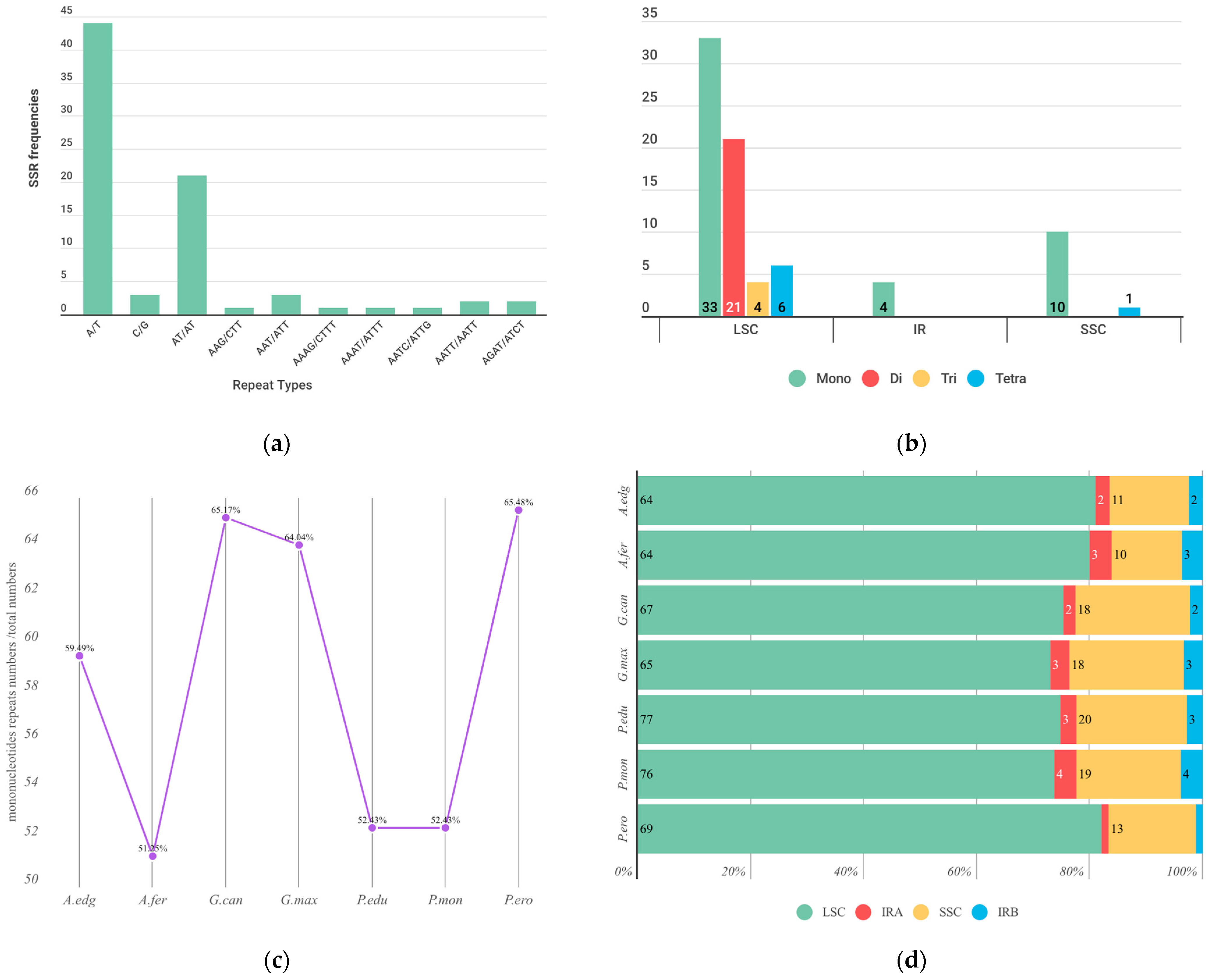
2.2. Repeat Analyses
2.3. Comparative Analysis of the Plastome in Subtribe Glycininae Species
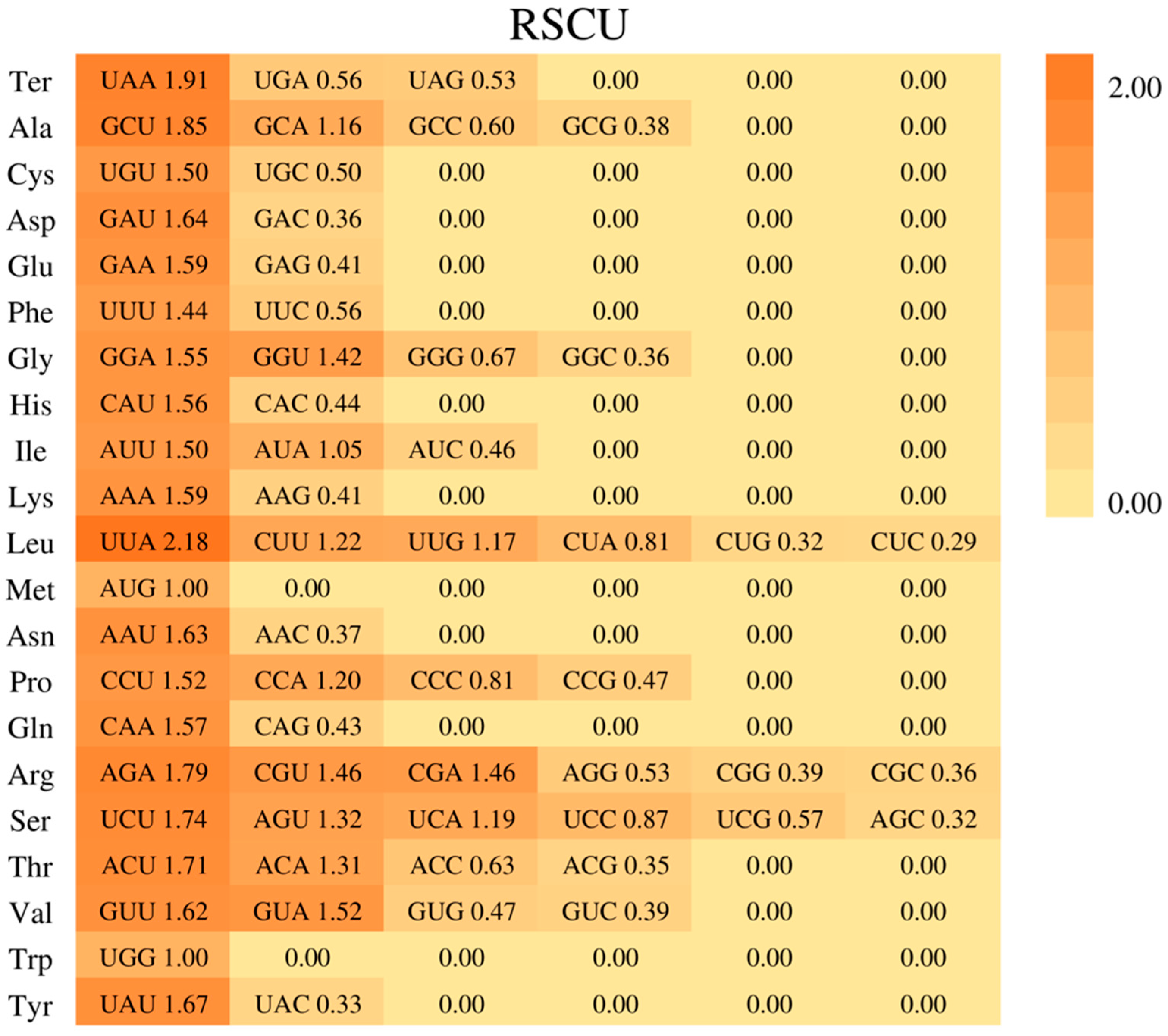
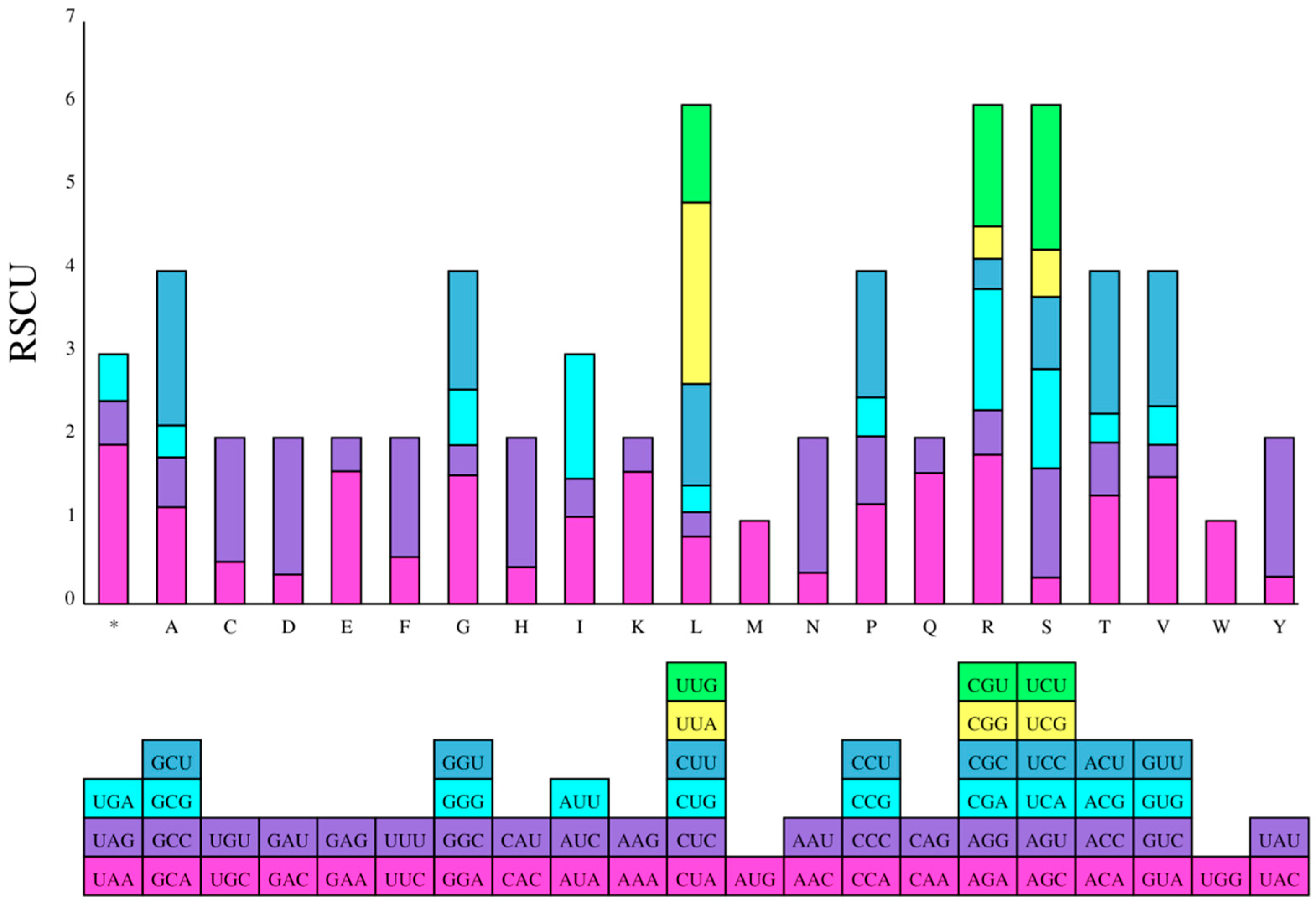
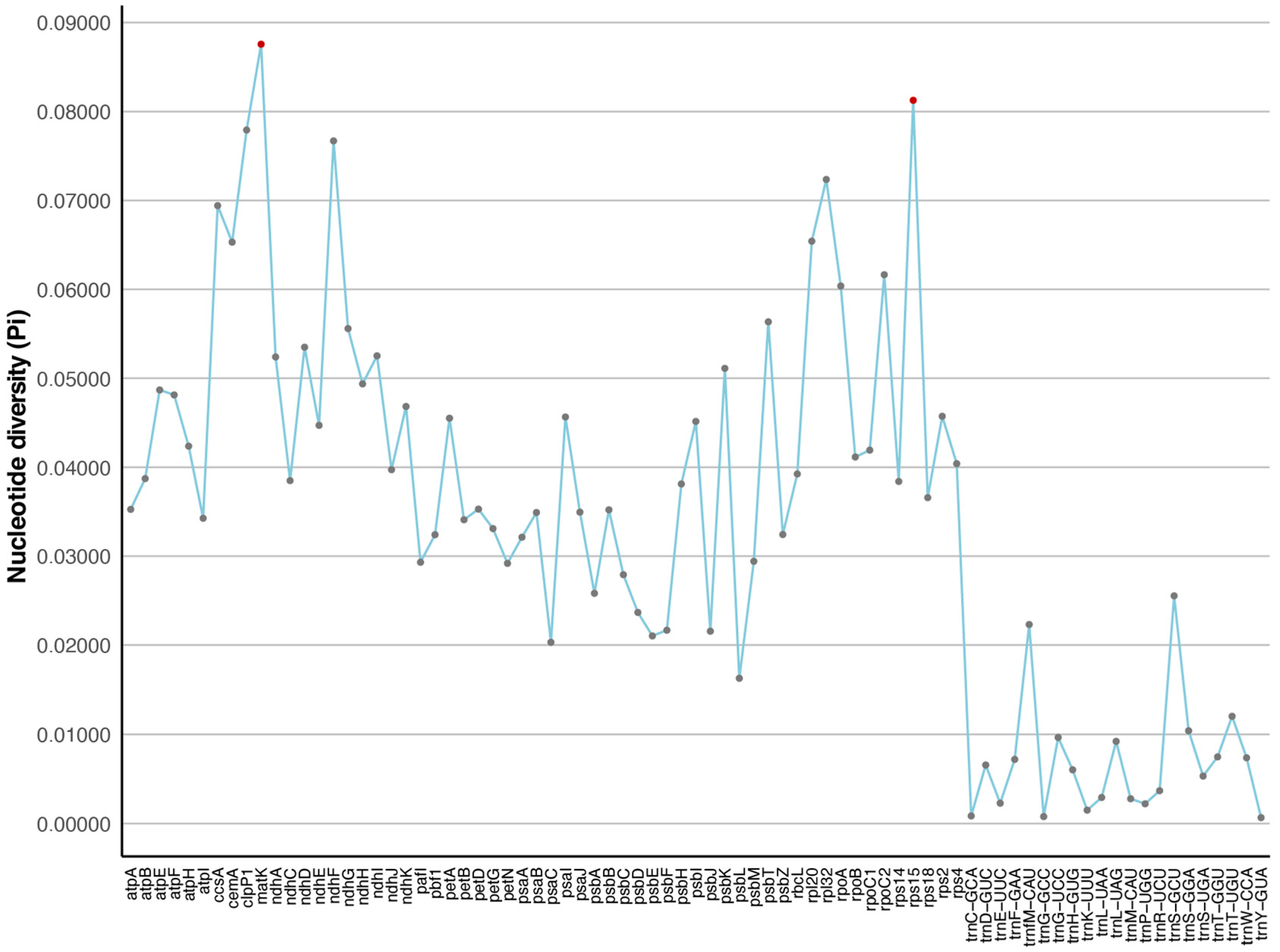
2.4. Divergence of Protein-Coding Gene Sequences
2.5. Phylogenetic Analysis
3. Materials and Methods
3.1. Plant Material and DNA Extraction
3.2. DNA Sequencing and Genome Assembly
3.3. Genome Annotation: Genes and Repetitive Elements
3.4. Genome Comparison and Phylogenetic Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Vasconcelos, M.W.; Grusak, M.A.; Pinto, E.; Gomes, A.; Ferreira, H.; Balázs, B.; Centofanti, T.; Ntatsi, G.; Savvas, D.; Karkanis, A. The Biology of Legumes and Their Agronomic, Economic, and Social Impact. In The Plant Family Fabaceae: Biology and Physiological Responses to Environmental Stresses; Springer: Berlin/Heidelberg, Germany, 2020; pp. 3–25. [Google Scholar]
- Ferreira, H.; Pinto, E.; Vasconcelos, M.W. Legumes as a Cornerstone of the Transition toward More Sustainable Agri-Food Systems and Diets in Europe. Front. Sustain. Food Syst. 2021, 5, 694121. [Google Scholar] [CrossRef]
- Didinger, C.; Thompson, H.J. The Role of Pulses in Improving Human Health: A Review. Legume Sci. 2022, 4, e147. [Google Scholar] [CrossRef]
- Aasfar, A.; Bargaz, A.; Yaakoubi, K.; Hilali, A.; Bennis, I.; Zeroual, Y.; Meftah Kadmiri, I. Nitrogen Fixing Azotobacter Species as Potential Soil Biological Enhancers for Crop Nutrition and Yield Stability. Front. Microbiol. 2021, 12, 628379. [Google Scholar] [CrossRef]
- Ahmad, E.; Zaidi, A.; Khan, M.S.; Oves, M. Heavy Metal Toxicity to Symbiotic Nitrogen-Fixing Microorganism and Host Legumes; Springer: Berlin/Heidelberg, Germany, 2012; ISBN 3-7091-0729-6. [Google Scholar]
- Gopalakrishnan, S.; Sathya, A.; Vijayabharathi, R.; Varshney, R.K.; Gowda, C.L.; Krishnamurthy, L. Plant Growth Promoting Rhizobia: Challenges and Opportunities. 3 Biotech 2015, 5, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Abusaief, H.M.A.-A.; Boasoul, S.H. A Taxonomic Study of Twelve Wild Forage Species of Fabaceae. Heliyon 2021, 7, e06077. [Google Scholar] [CrossRef]
- Cardoso, D.; de Queiroz, L.P.; Pennington, R.T.; de Lima, H.C.; Fonty, E.; Wojciechowski, M.F.; Lavin, M. Revisiting the Phylogeny of Papilionoid Legumes: New Insights from Comprehensively Sampled Early-Branching Lineages. Am. J. Bot. 2012, 99, 1991–2013. [Google Scholar] [CrossRef]
- Group, T.L.P.W.; Bruneau, A.; Doyle, J.J.; Herendeen, P.; Hughes, C.; Kenicer, G.; Lewis, G.; Mackinder, B.; Pennington, R.T.; Sanderson, M.J.; et al. Legume Phylogeny and Classification in the 21st Century: Progress, Prospects and Lessons for Other Species–Rich Clades. TAXON 2013, 62, 217–248. [Google Scholar] [CrossRef]
- Melgar, A.E.; Zelada, A.M. Evolutionary Analysis of Angiosperm Dehydrin Gene Family Reveals Three Orthologues Groups Associated to Specific Protein Domains. Sci. Rep. 2021, 11, 23869. [Google Scholar] [CrossRef]
- Wojciechowski, M.F. Towards a New Classification of Leguminosae: Naming Clades Using Non-Linnaean Phylogenetic Nomenclature. S. Afr. J. Bot. 2013, 89, 85–93. [Google Scholar] [CrossRef]
- Lackey, J.A. A Revised Classification of the Tribe Phaseoleae (Leguminosae: Papilionoideae), and Its Relation to Canavanine Distribution. Bot. J. Linn. Soc. 1977, 74, 163–178. [Google Scholar] [CrossRef]
- Li, S. Phaseoleae. In Flora Reipublicae Popularis Sinicae; Wu, C.Y., Ed.; Science Press: Beijing, China, 1995; Volume 41, ISBN 7-03-004440-1. [Google Scholar]
- Oyebanji, O.; Zhang, R.; Chen, S.-Y.; Yi, T. New Insights Into the Plastome Evolution of the Millettioid/Phaseoloid Clade (Papilionoideae, Leguminosae). Front. Plant Sci. 2020, 11, 151. [Google Scholar] [CrossRef]
- Cheplick, G.P. Life History Evolution in Amphicarpic Plants. Plant Species Biol. 1994, 9, 119–131. [Google Scholar] [CrossRef]
- Schnee, B.K.; Waller, D.M. Reproductive Behavior of Amphicarpaea bracteata (Leguminosae), an Amphicarpic Annual. Am. J. Bot. 1986, 73, 376–386. [Google Scholar] [CrossRef]
- Cheplick, G.P. The Ecology of Amphicarpic Plants. Trends Ecol. Evol. 1987, 2, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Baskin, J.M.; Baskin, C.C.; Cheplick, G.P.; Yang, X.; Huang, Z. Amphicarpic Plants: Definition, Ecology, Geographic Distribution, Systematics, Life History, Evolution and Use in Agriculture. Biol. Rev. 2020, 95, 1442–1466. [Google Scholar] [CrossRef]
- Song, T.; Zhou, M.; Yuan, Y.; Yu, J.; Cai, H.; Li, J.; Chen, Y.; Bai, Y.; Zhou, G.; Cui, G. Chromosome-Scale Reference Genome of Amphicarpaea Edgeworthii: A New Resource for Amphicarpic Plants Research and Complex Flowering Pattern. Front. Plant Sci. 2021, 12, 770660. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Zhou, M.; Yuan, Y.; Yu, J.; Cai, H.; Li, J.; Chen, Y.; Bai, Y.; Zhou, G.; Cui, G. First High-Quality Reference Genome of Amphicarpaea edgeworthii. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Han, K.; Li, R.; Xu, G.; Han, Y.; Cui, F.; Fan, S.; Seim, I.; Fan, G.; et al. Insights into Amphicarpy from the Compact Genome of the Legume Amphicarpaea edgeworthii. Plant Biotechnol. J. 2021, 19, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhao, Z.-N.; Ping, H.-L. The Complete Plastid Genome of Amphicarpaea ferruginea Bentham (Leguminosae), a Grass Species with Development and Utilization Prospect. Mitochondrial DNA B Resour. 2022, 7, 1221–1223. [Google Scholar] [CrossRef]
- Lee, J.; Hymowitz, T. A Molecular Phylogenetic Study of the Subtribe Glycininae (Leguminosae) Derived from the Chloroplast DNA Rps16 Intron Sequences. Am. J. Bot. 2001, 88, 2064–2073. [Google Scholar] [CrossRef]
- Agyei, D. Bioactive Proteins and Peptides from Soybeans. Recent Pat. Food Nutr. Agric. 2015, 7, 100–107. [Google Scholar] [CrossRef]
- Rackis, J.J. Biological and Physiological Factors in Soybeans. J. Am. Oil Chem. Soc. 1974, 51, 161A–174A. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, W.; Xiu, L.; Sun, Y.; Gu, W.; Wang, Y.; Zhang, H.; Chen, W. Soybean Yield Response of Biochar-Regulated Soil Properties and Root Growth Strategy. Agronomy 2022, 12, 1412. [Google Scholar] [CrossRef]
- Kirchhoff, H. Chloroplast Ultrastructure in Plants. New Phytol. 2019, 223, 565–574. [Google Scholar] [CrossRef]
- Ozeki, H.; Umesono, K.; Inokuchi, H.; Kohchi, T.; Ohyama, K. The Chloroplast Genome of Plants: A Unique Origin. Genome 1989, 31, 169–174. [Google Scholar] [CrossRef]
- Yang, Q.; Jiang, Y.; Wang, Y.; Han, R.; Liang, Z.; He, Q.; Jia, Q. SSR Loci Analysis in Transcriptome and Molecular Marker Development in Polygonatum sibiricum. Biomed Res Int 2022, 2022, 4237913. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, M.-M.; Hu, X.-F.; Wang, R.-H.; He, Q.-L. The Complete Chloroplast Genome Sequence of Spiraea japonica var. acuminata Franch. (Rosaceae). Mitochondrial DNA B Resour. 2022, 7, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Li, Y.; Chen, X.; Yan, Y.; Xia, P. Characterization of the Complete Chloroplast Genome Sequence of Elaeagnus henryi Warb. Ex Diels (Elaeagnaceae). Mitochondrial DNA B Resour. 2022, 7, 1876–1878. [Google Scholar] [CrossRef]
- Li, Q.; Chen, X.; Yang, D.; Xia, P. Genetic Relationship of Pleione Based on the Chloroplast Genome. Gene 2023, 858, 147203. [Google Scholar] [CrossRef]
- Guo, L.; Wang, X.; Wang, R.; Li, P. Characterization and Comparative Analysis of Chloroplast Genomes of Medicinal Herb Scrophularia ningpoensis and Its Common Adulterants (Scrophulariaceae). Int. J. Mol. Sci. 2023, 24, 10034. [Google Scholar] [CrossRef]
- Wang, R.; Gao, J.; Feng, J.; Yang, Z.; Qi, Z.; Li, P.; Fu, C. Comparative and Phylogenetic Analyses of Complete Chloroplast Genomes of Scrophularia incisa Complex (Scrophulariaceae). Genes 2022, 13, 1691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, M.-M.; Zhang, X.-M.; Chen, S.-N.; Liang, Z.-S. The Complete Chloroplast Genome Sequence of Traditional Chinese Medicine uncaria Macrophylla (Rubiaceae). Mitochondrial DNA B Resour. 2022, 7, 694–695. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yao, X.; Zhang, C.; Xia, P. Characterization of the Complete Chloroplast Genome Sequence of Cardamine lyrata Bunge (Brassicaceae). Mitochondrial DNA B Resour. 2022, 7, 936–937. [Google Scholar] [CrossRef]
- Chen, M.-M.; Zhang, M.; Liang, Z.-S.; He, Q.-L. Characterization and Comparative Analysis of Chloroplast Genomes in Five Uncaria Species Endemic to China. Int. J. Mol. Sci. 2022, 23, 11617. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-M.; Wang, F.; Yan, S.-Y.; Zhu, Z.-M.; Gao, X.-F.; Zhao, X.-L. Phylogenomics and Plastome Evolution of Indigofera (Fabaceae). Front. Plant Sci. 2023, 14, 1186598. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wu, L.; Wang, Q.; Pan, Y.; Li, B.; Lin, Y.; Yao, H. Comparison Analysis Based on Complete Chloroplast Genomes and Insights into Plastid Phylogenomic of Four Iris Species. BioMed Res. Int. 2022, 2022, e2194021. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.-H.; Jin, J.-J.; Stull, G.W.; Bruneau, A.; Cardoso, D.; De Queiroz, L.P.; Moore, M.J.; Zhang, S.-D.; Chen, S.-Y.; et al. Exploration of Plastid Phylogenomic Conflict Yields New Insights into the Deep Relationships of Leguminosae. Syst. Biol. 2020, 69, 613–622. [Google Scholar] [CrossRef]
- Choi, I.-S.; Cardoso, D.; de Queiroz, L.P.; de Lima, H.C.; Lee, C.; Ruhlman, T.A.; Jansen, R.K.; Wojciechowski, M.F. Highly Resolved Papilionoid Legume Phylogeny Based on Plastid Phylogenomics. Front. Plant Sci. 2022, 13, 823190. [Google Scholar] [CrossRef]
- Moghaddam, M.; Ohta, A.; Shimizu, M.; Terauchi, R.; Kazempour-Osaloo, S. The Complete Chloroplast Genome of Onobrychis Gaubae (Fabaceae-Papilionoideae): Comparative Analysis with Related IR-Lacking Clade Species. BMC Plant Biol. 2022, 22, 75. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Xiong, Y.; He, J.; Yu, Q.; Zhao, J.; Lei, X.; Dong, Z.; Yang, J.; Peng, Y.; Zhang, X. The Complete Chloroplast Genome of Two Important Annual Clover Species, Trifolium Alexandrinum and T. Resupinatum: Genome Structure, Comparative Analyses and Phylogenetic Relationships with Relatives in Leguminosae. Plants 2020, 9, 478. [Google Scholar] [CrossRef] [PubMed]
- Lackey, J.A. Cotyledon Areoles in Subtribe Kennediinae (Leguminosae: Phaseoleae). Aust. J. Bot. 2008, 56, 265–271. [Google Scholar] [CrossRef]
- Bruneau, A. Phylogenetic and Biogeographical Patterns in Erythrina (Leguminosae: Phaseoleae) as Inferred from Morphological and Chloroplast DNA Characters. Syst. Bot. 1996, 24, 587–605. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality Control and Preprocessing of Metagenomic Datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De Novo Assembly of Organelle Genomes from Whole Genome Data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an Integrated Plastome Sequence Annotator and Analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3. 1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST Databases for the Development and Characterization of Gene-Derived SSR-Markers in Barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The Manifold Applications of Repeat Analysis on a Genomic Scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing Global DNA Sequence Alignments of Arbitrary Length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree V1. 3.1: Tree Figure Drawing Tool. Available online: http://treebio.ed.ac.uk/software/figtree/ (accessed on 1 May 2023).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | A (%) | T (%) | G (%) | C (%) | GC (%) | Total (bp) | Proportion in Genome (%) |
|---|---|---|---|---|---|---|---|
| Genome | 32.3 | 32.3 | 17.8 | 17.6 | 35.4 | 148,650 | 100 |
| CDS | 31.7 | 32.3 | 19.3 | 16.7 | 36.0 | 75,693 | 50.92 |
| tRNA | 34.0 | 32.6 | 18.4 | 15.2 | 33.5 | 22,185 | 14.92 |
| rRNA | 21.6 | 24.9 | 29.9 | 23.6 | 53.6 | 2543 | 1.71 |
| Cis-spliced intron | 26.2 | 18.9 | 31.5 | 23.5 | 54.9 | 9060 | 6.09 |
| Non-coding region | 35.1 | 34.7 | 15.1 | 15.1 | 30.2 | 49,351 | 33.20 |
| LSC | 33.6 | 33.5 | 16.8 | 16.0 | 32.9 | 83,675 | 56.29 |
| SSC | 35.5 | 35.8 | 13.5 | 15.2 | 28.7 | 17,967 | 12.10 |
| IRA | 28.6 | 29.1 | 21.9 | 20.4 | 42.3 | 23,504 | 15.82 |
| IRB | 28.6 | 29.1 | 21.9 | 20.4 | 42.3 | 23,504 | 15.82 |
| Category | Group of Genes | Name of Genes |
|---|---|---|
| RNA genes | Ribosomal RNA genes (rRNA) | rrn5 a, rrn4.5 a, rrn16 a, rrn23 a |
| Transfer RNA genes (tRNA) | trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC, trnH-GUG, trnI-GAU a, trnK-UUU, trnL-CAA a, trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU a, trnP-UGG, trnQ-UUG, trnR-ACG a, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC a, trnV-UAC, trnW-CCA, trnY-GUA, trnA-UGC a | |
| Ribosomal proteins | Small subunit of ribosome | rps11, rps14, rps15, rps16 +, rps18, rps2, rps3, rps4, rps7 a, rps8, rps12 +,a, rps19 |
| Transcription | Large subunit of ribosome | rpl14, rpl16, rpl2 +, rpl20, rpl22, rpl23 a, rpl32, rpl33, rpl36 |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1 +, rpoC2 | |
| Protein genes Other genes | Photosystem I | psaA, psaB, psaC, psaI, psaJ, pafI ++, pafII |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbT, psbZ | |
| Subunit of cytochrome | petA, petB +, petD +, petG, petL, petN | |
| Subunit of ATP synthase | atpA, atpB, atpE, atpF +, atpH, atpI | |
| Chloroplast envelope membrabe protien | cemA | |
| NADH dehydrogenase | ndhA +, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Large subunit of Rubisco | rbcL | |
| Subunit acetyl-coA carboxylase | accD | |
| ATP-dependent protease subunit P | clpP1 ++ | |
| Maturase | matK | |
| C-type cytochrome synthesis | ccsA | |
| Component of the TIC complex | ycf1 a | |
| Hypothetical proteins | ycf2 a | |
| Translation initiation factor | infA | |
| N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein | pbf1 |
| Gene | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|
| ndhA | SSC | 551 | 1273 | 541 | ||
| trnA-UGC | IR | 38 | 807 | 35 | ||
| trnI-GAU | IR | 42 | 944 | 37 | ||
| rps12 | IR | 114 | 535 | 232 | ||
| rpl2 | IR | 391 | 719 | 434 | ||
| rpl16 | LSC | 9 | 1176 | 399 | ||
| petD | LSC | 8 | 729 | 475 | ||
| petB | LSC | 6 | 812 | 642 | ||
| clpP1 | LSC | 71 | 708 | 292 | 790 | 228 |
| rps16 | LSC | 40 | 879 | 230 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, Y.-N.; Wang, X.-Q.; Ding, L.-L.; Bai, X.-Y.; Feng, Y.-Q.; Qi, Z.-C.; Sun, Y.-T.; Yan, X.-L. Deciphering the Plastomic Code of Chinese Hog-Peanut (Amphicarpaea edgeworthii Benth., Leguminosae): Comparative Genomics and Evolutionary Insights within the Phaseoleae Tribe. Genes 2024, 15, 88. https://doi.org/10.3390/genes15010088
Xiang Y-N, Wang X-Q, Ding L-L, Bai X-Y, Feng Y-Q, Qi Z-C, Sun Y-T, Yan X-L. Deciphering the Plastomic Code of Chinese Hog-Peanut (Amphicarpaea edgeworthii Benth., Leguminosae): Comparative Genomics and Evolutionary Insights within the Phaseoleae Tribe. Genes. 2024; 15(1):88. https://doi.org/10.3390/genes15010088
Chicago/Turabian StyleXiang, Yi-Nan, Xiao-Qun Wang, Lu-Lu Ding, Xin-Yu Bai, Yu-Qing Feng, Zhe-Chen Qi, Yong-Tao Sun, and Xiao-Ling Yan. 2024. "Deciphering the Plastomic Code of Chinese Hog-Peanut (Amphicarpaea edgeworthii Benth., Leguminosae): Comparative Genomics and Evolutionary Insights within the Phaseoleae Tribe" Genes 15, no. 1: 88. https://doi.org/10.3390/genes15010088