
Genesis, Evolution, and Genetic Diversity of the Hexaploid, Narrow Endemic Centaurea tentudaica

 , ,
, ,
Abstract
:1. Introduction
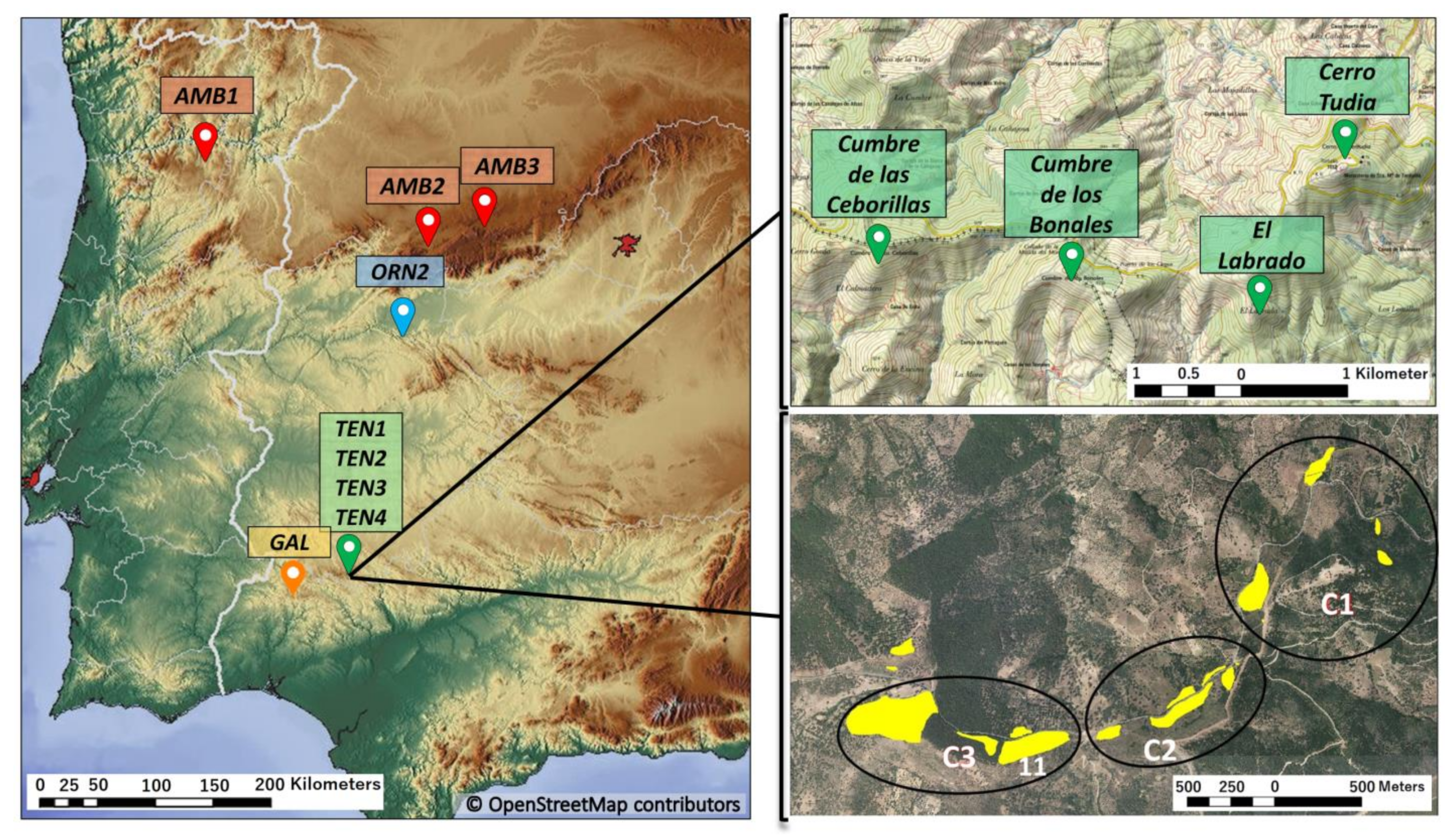
2. Materials and Methods
2.1. Sampling Strategy and Markers
2.2. DNA Amplification, Cloning, and Sequencing
2.3. Sequence Alignment and Phylogenetic Analyses
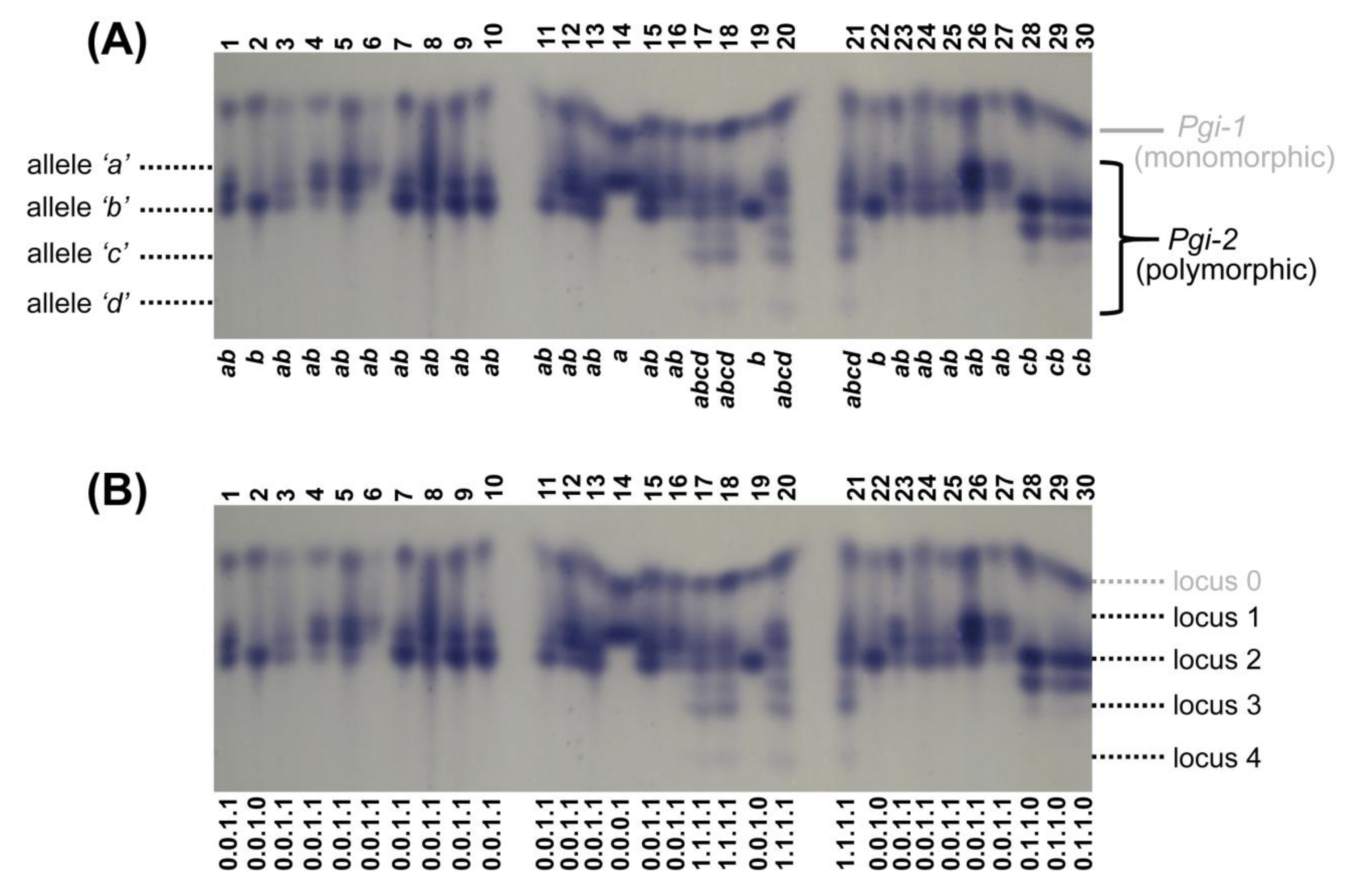
2.4. Allozyme Extraction and Analyses
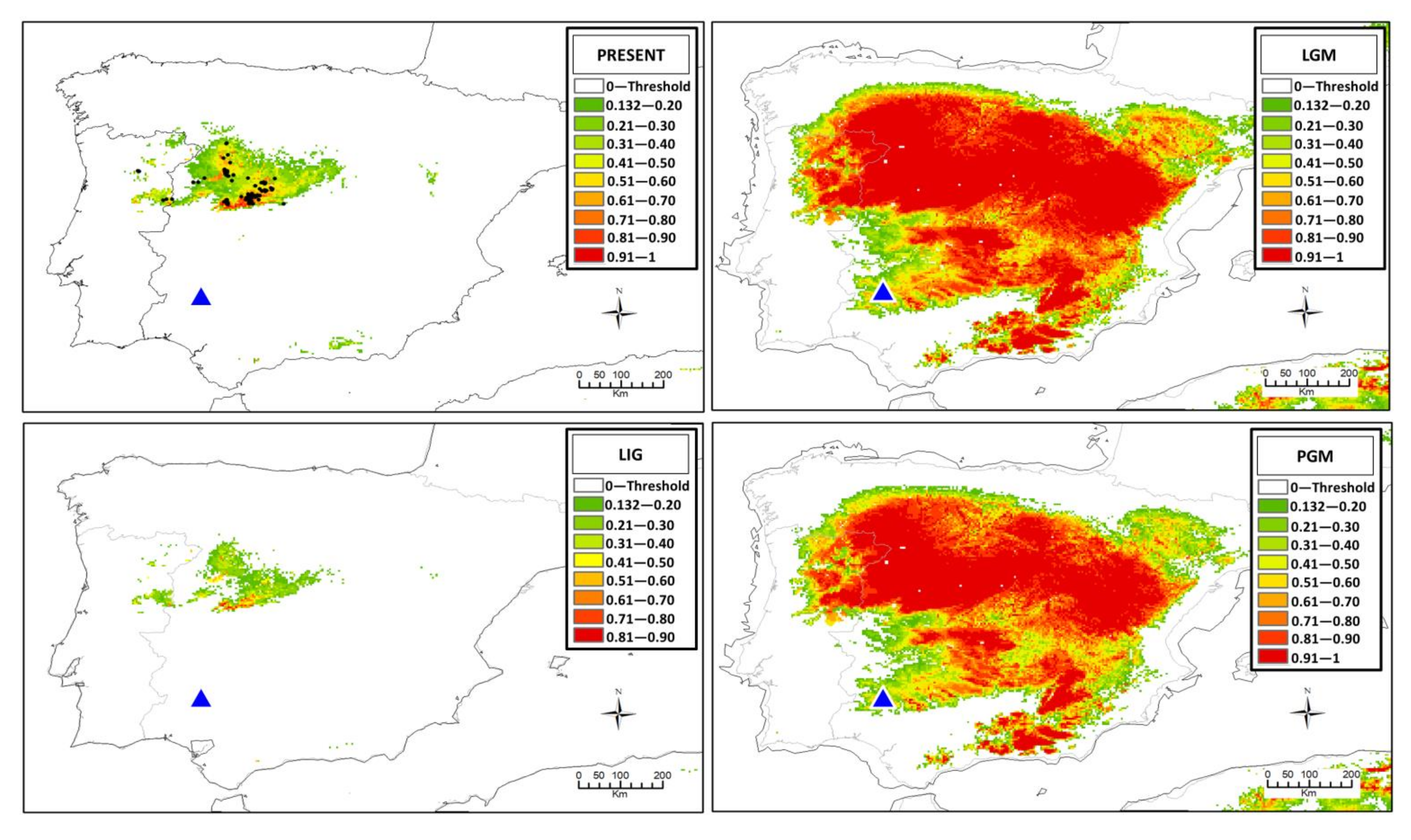
2.5. Ecological Niche Modeling
3. Results
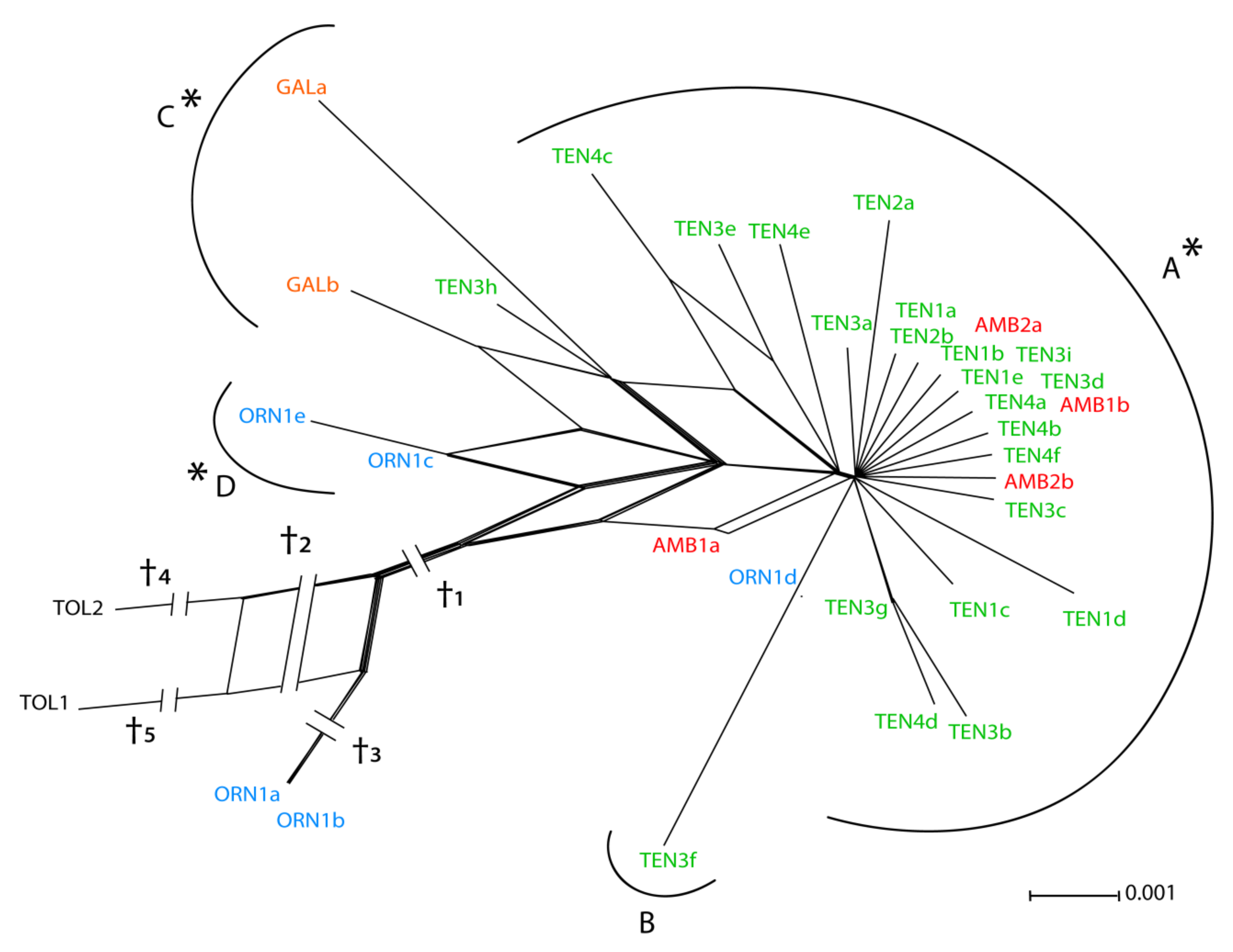
3.1. ETS Region
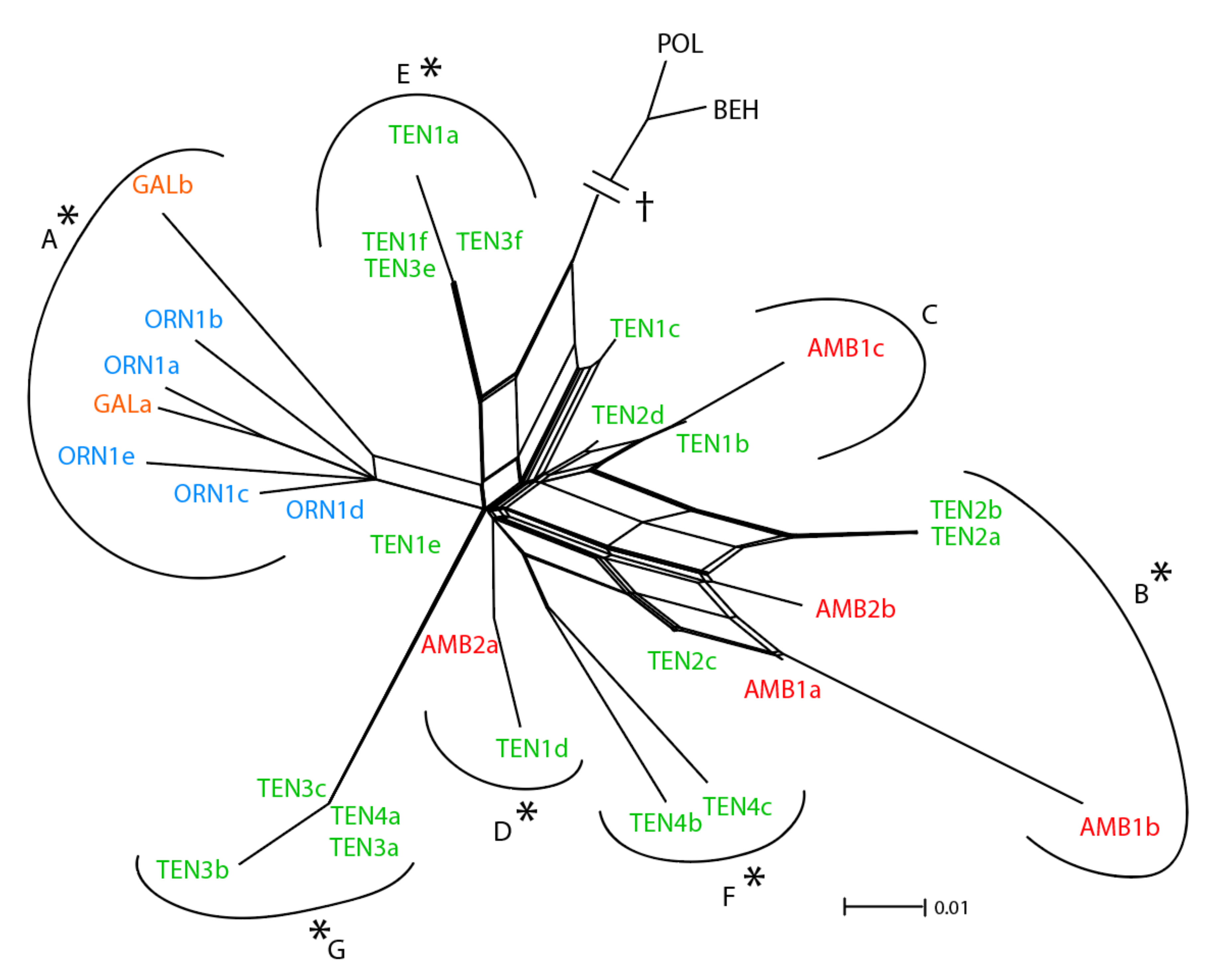
3.2. Low-copy Genes AGT1 and PgiC
3.3. Allozymes
3.4. Ecological Niche Modeling
4. Discussion
4.1. Relative Utility of ETS, AGT1, and PgiC in Disentangling the Origin of Centaurea tentudaica
4.2. The Origin of Centaurea tentudaica
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mallet, J. Hybrid speciation. Nature 2007, 446, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Soltis, P.S.; Marchant, D.B.; Van de Peer, Y.; Soltis, D.E. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 2015, 35, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Landis, J.B.; Soltis, D.E.; Li, Z.; Marx, H.E.; Barker, M.S.; Tank, D.C.; Soltis, P.S. Impact of whole-genome duplication events on diversification rates in angiosperms. Am. J. Bot. 2018, 105, 348–363. [Google Scholar] [CrossRef] [Green Version]
- Soltis, P.S.; Soltis, D.E. The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 2009, 60, 561–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, E. Systematics and the Origin of Species; Columbia University Press: New York, NY, USA, 1942. [Google Scholar]
- De Queiroz, K. Species concepts and species delimitation. Syst. Biol. 2007, 56, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Brennan, A.C.; Barker, D.; Hiscock, S.J.; Abbott, R.J. Molecular genetic and quantitative trait divergence associated with recent homoploid hybrid speciation: A study of Senecio squalidus (Asteraceae). Heredity 2012, 108, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Mameli, G.; López-Alvarado, J.; Farris, E.; Susanna, A.; Filigheddu, R.; Garcia-Jacas, N. The role of parental and hybrid species in multiple introgression events: Evidence of homoploid hybrid speciation in Centaurea (Cardueae, Asteraceae). Bot. J. Linn. Soc. 2014, 175, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Jacas, N.; Soltis, P.S.; Font, M.; Soltis, D.E.; Vilatersana, R.; Susanna, A. The polyploid series of Centaurea toletana: Glacial migrations and introgression revealed by nrDNA and cpDNA sequence analyzes. Mol. Phylogenet. Evol. 2009, 52, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Linder, C.R.; Goertzen, L.R.; Heuvel, B.V.; Francisco-Ortega, J.; Jansen, R.K. The complete external transcribed spacer of 18S-26S rDNA: Amplification and phylogenetic utility at low taxonomic levels in Asteraceae and closely allied families. Mol. Phylogenet. Evol. 2000, 14, 285–303. [Google Scholar] [CrossRef]
- Font, M.; Vallès, J.; Susanna, A.; Garcia-Jacas, N. Auto- and allopolyploidy in Centaurea sect. Acrocentron s. l. (Asteraceae, Cardueae): Karyotype and fluorochrome banding pattern analyses. Collect. Bot. 2008, 27, 7–18. [Google Scholar]
- Font, M.; Garcia-Jacas, N.; Vilatersana, R.; Roquet, C.; Susanna, A. Evolution and biogeography of Centaurea section Acrocentron inferred from nuclear and plastid DNA sequence analyses. Ann. Bot. 2009, 103, 985–997. [Google Scholar] [CrossRef]
- Small, R.L.; Cronn, R.C.; Wendel, J.F. Use of nuclear genes for phylogeny reconstruction in plants. Aust. Syst. Bot. 2004, 17, 145–170. [Google Scholar] [CrossRef]
- Ford, V.S.; Lee, J.; Baldwin, B.G.; Gottlieb, L.D. Species divergence and relationships in Stephanomeria (Compositae): PgiC phylogeny compared to prior biosystematic studies. Am. J. Bot. 2006, 93, 480–490. [Google Scholar] [CrossRef]
- López-Pujol, J.; Bosch, M.; Simon, J.; Blanché, C. Allozyme diversity in the tetraploid endemic Thymus loscosii (Lamiaceae). Ann. Bot. 2004, 93, 323–332. [Google Scholar] [CrossRef]
- Palop-Esteban, M.; Segarra-Moragues, J.G.; González-Candelas, F. Polyploid origin, genetic diversity and population structure in the tetraploid sea lavender Limonium narbonense Miller (Plumbaginaceae) from eastern Spain. Genetica 2011, 139, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Hamrick, J.L.; Godt, M.J.W. Allozyme diversity in plant species. In Plant Population Genetics, Breeding, and Genetic Resources; Brown, A.H.D., Clegg, M.T., Kahler, A.L., Weir, B.S., Eds.; Sinauer Associates: Sunderland, MA, USA, 1990; pp. 43–63. [Google Scholar]
- Hewitt, G.M. Mediterranean peninsulas: The evolution of hotspots. In Biodiversity Hotspots; Zachos, F.E., Habel, J.C., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 123–147. [Google Scholar]
- Peñalba, M.C.; Arnold, M.; Guiot, J.; Duplessy, J.C.; de Beaulieu, J.L. Termination of the last glaciation in the Iberian Peninsula inferred from the pollen sequence of Quintanar de la Sierra. Quat. Res. 1997, 48, 205–214. [Google Scholar] [CrossRef]
- Hewitt, G.M. Quaternary phylogeography: The roots of hybrid zones. Genetica 2011, 139, 617–638. [Google Scholar] [CrossRef] [PubMed]
- López-Alvarez, D.; Manzaneda, A.J.; Rey, P.J.; Giraldo, P.; Benavente, E.; Allainguillaume, J.; Mur, L.; Caicedo, A.L.; Hazen, S.P.; Breiman, A.; et al. Environmental niche variation and evolutionary diversification of the Brachypodium distachyon grass complex species in their native circum-Mediterranean range. Am. J. Bot. 2015, 102, 1073–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, I.; Draper, D.; López-Herranz, M.L.; Garnatje, T.; Segarra-Moragues, J.G.; Catalán, P. Past climate changes facilitated homoploid speciation in three mountain spiny fescues (Festuca, Poaceae). Sci. Rep. 2016, 6, 36283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Luis, M.; Álvarez-Jiménez, J.; Rejos, F.J.; Bartolomé, C. Using species distribution models to locate the potential cradles of the allopolyploid Gypsophila bermejoi G. López (Caryophyllaceae). PLoS ONE 2020, 15, e0232736. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Jacas, N.; Susanna, A. Centaurea prolongi and C. crocata in Portugal: An old confusion. Nord. J. Bot. 1994, 14, 31–38. [Google Scholar] [CrossRef]
- Garcia-Jacas, N. Centaurea kunkelii (Asteraceae, Cardueae), a new hybridogenic endecaploid species of sect. Acrocentron from Spain. Ann. Bot. Fenn. 1998, 35, 159–167. [Google Scholar]
- Font, M.; Garnatje, T.; Garcia-Jacas, N.; Susanna, A. Delineation and phylogeny of Centaurea sect. Acrocentron based on DNA sequences: A restoration of the genus Crocodylium and indirect evidence of introgression. Plant Syst. Evol. 2002, 234, 15–26. [Google Scholar] [CrossRef]
- Hilpold, A.; Garcia-Jacas, N.; Vilatersana, R.; Susanna, A. Taxonomical and nomenclatural notes on Centaurea: A proposal of classification, a description of new sections and subsections, and a species list of the redefined section Centaurea. Collect. Bot. 2014, 33, e001. [Google Scholar] [CrossRef] [Green Version]
- Márquez, F. Centaurea de Tentudía: Estudios Para su Conservación. Ph.D. Thesis, University of Extremadura, Badajoz, Spain, 2015. [Google Scholar]
- Rivas Goday, S. Vegetación y Flórula de la Cuenca Extremeña del Guadiana (Vegetación y Flórula de la Provincia de Badajoz); Publicaciones de la Excma. Diputación Provincial de Badajoz: Madrid, Spain, 1964. [Google Scholar]
- Rivas Martínez, S. De nomenclatura notulae, I. Lazaroa 1980, 2, 327–328. [Google Scholar]
- Fernández Casas, F.J.; Susanna, A. De Centaureis occidentalibus notulae sparsae III. Fontqueria 1982, 1, 1–8. [Google Scholar]
- Rivas Martínez, S. Bioclimatología, biogeografía y series de vegetación de Andalucía Occidental. Lagascalia 1988, 15, 91–119. [Google Scholar]
- Márquez, F.; Alonso, D.G.; Vázquez, F.M. Estudio de distribución y caracterización del hábitat del taxon amenazado Centaurea amblensis subsp. tentudaica (Rivas Goday) Rivas-Martínez. Folia Bot. Extremad. 2011, 5, 37–43. [Google Scholar]
- Fernández Casas, F.J.; Susanna, A. Monografía de la sección Chamaecyanus Willk. del género Centaurea L. Treb. Inst. Bot. Barc. 1985, 10, 1–174. [Google Scholar]
- Soltis, D.E.; Soltis, P.S.; Schemske, D.W.; Hancock, J.F.; Thompson, J.N.; Husband, B.C.; Judd, W.S. 2007. Autopolyploidy in angiosperms: Have we grossly underestimated the number of species? Taxon 2007, 56, 13–30. [Google Scholar]
- López-Pujol, J.; Garcia-Jacas, N.; Susanna, A.; Vilatersana, R. Should we conserve pure species or hybrid species? Delimiting hybridization and introgression in the Iberian endemic Centaurea podospermifolia. Biol. Conserv. 2012, 152, 271–279. [Google Scholar] [CrossRef]
- Doyle, J.J.; Dickson, E.E. Preservation of plant samples for DNA restriction endonuclease analysis. Taxon 1987, 36, 715–722. [Google Scholar] [CrossRef]
- Cullings, K.W. Design and testing of a plant-specific PCR primer for ecological and evolutionary studies. Mol. Ecol. 1992, 1, 233–240. [Google Scholar] [CrossRef]
- Tel-Zur, N.; Abbo, S.; Mysladbodksi, D.; Mizrahi, Y. Modified CTAB procedure for DNA isolation from epiphytic cacti of genera Hylocereus and Selenicereus (Cactaceae). Plant Mol. Biol. Rep. 1999, 17, 249–254. [Google Scholar] [CrossRef]
- Baldwin, B.G.; Markos, S. Phylogenetic utility of the External Transcribed Spacer (ETS) of 18S–26S rDNA: Congruence of ETS and ITS trees of Calycadenia (Compositae). Mol. Phylogenet. Evol. 1998, 10, 449–463. [Google Scholar] [CrossRef]
- Susanna, A.; Galbany-Casals, M.; Romaschenko, K.; Barres, L.; Martín, J.; Garcia-Jacas, N. Lessons from Plectocephalus (Compositae, Cardueae-Centaureinae): ITS disorientation in annuals and Beringian dispersal revealed by molecular analyses. Ann. Bot. 2011, 108, 263–277. [Google Scholar] [CrossRef]
- Li, M.; Wunder, J.; Bissoli, G.; Scarponi, E.; Gazzani, S.; Barbaro, E.; Saedler, H.; Varotto, C. Development of COS genes as universally amplifiable markers for phylogenetic reconstructions of closely related plant species. Cladistics 2008, 24, 727–745. [Google Scholar] [CrossRef] [Green Version]
- Vilatersana, R.; Brysting, A.K.; Brochmann, C. Molecular evidence for hybrid origins of the invasive polyploids Carthamus creticus and C. turkestanicus (Cardueae, Asteraceae). Mol. Phylogenet. Evol. 2007, 44, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Cline, J.; Braman, J.C.; Hogrefe, H.H. PCR fidelity of Pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996, 24, 3546–3551. [Google Scholar] [CrossRef] [Green Version]
- Popp, M.; Oxelman, B. Inferring the history of the polyploid Silene aegaea (Caryophyllaceae) using plastid and homoeologous nuclear DNA sequences. Mol. Phylogenet. Evol. 2001, 20, 474–481. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; Van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Wendel, F.; Weeden, N.F. Visualization and interpretation of plant isozymes. In Isozymes in Plant Biology; Soltis, D.E., Soltis, P.S., Eds.; Springer: Dordrecht, The Netherlands, 1989; pp. 5–45. [Google Scholar]
- Rogers, D.L. Genotypic diversity and clone size in old-growth populations of coast redwood (Sequoia sempervirens). Can. J. Bot. 2000, 78, 1408–1419. [Google Scholar]
- Wang, A.; Chen, Y.; Chen, G.; Lee, J.; Fu, C. Relationships and hybridization among Smilax china and its affinities: Evidence from allozyme data. Biochem. Genet. 2008, 46, 281–292. [Google Scholar] [CrossRef]
- López-Vinyallonga, S.; Soriano, I.; Susanna, A.; Montserrat, J.M.; Roquet, C.; Garcia-Jacas, N. The polyploid series of the Achillea millefolium aggregate in the Iberian Peninsula investigated using microsatellites. PLoS ONE 2015, 10, e0129861. [Google Scholar]
- Zozomová-Lihová, J.; Krak, K.; Mandákova, T.; Shimizu, K.K.; Španiel, S.; Vít, P.; Lysak, M.A. Multiple hybridization events in Cardamine (Brassicaceae) during the last 150 years: Revisiting a textbook example of neoallopolyploidy. Ann. Bot. 2014, 113, 817–830. [Google Scholar] [CrossRef] [Green Version]
- Swofford, D.L.; Selander, R.B. Biosys-1: Release 1.7. A Computer Program for the Analysis of Allelic Variation in Genetics. User’s Manual; Department of Genetics and Development, University of Illinois: Urbana-Champaign, IL, USA, 1989. [Google Scholar]
- Vekemans, X. AFLP-SURV Ver. 1.0 A Program for Genetic Diversity Analysis with AFLP (and RAPD) Population Data; Distributed by the author; Laboratoire de Génétique et Ecologie Végétale, Université Libre de Bruxelles: Brussels, Belgium, 2002. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Pearson, R.G.; Raxworthy, C.J.; Nakamura, M.; Peterson, A.T. Predicting species distributions from small numbers of occurrence records: A test case using cryptic geckos in Madagascar. J. Biogeogr. 2007, 34, 102–117. [Google Scholar] [CrossRef]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.J.; Dudík, M. Modeling of species distributions with MaxEnt: New extensions and a comprehensive evaluation. Ecography 2008, 31, 161–175. [Google Scholar] [CrossRef]
- Brown, J.L.; Bennett, J.R.; French, C.M. SDMtoolbox 2.0: The next generation Python-based GIS toolkit for landscape genetic, biogeographic and species distribution model analyses. PeerJ 2017, 5, e4095. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, Y.; Lambeck, K.; Deckker, P.D.; Johnston, P.; Fifield, L.K. Timing of the Last Glacial Maximum from observed sea-level minima. Nature 2000, 406, 713–716. [Google Scholar] [CrossRef]
- Huybrechts, P. Sea-level changes at the LGM from ice-dynamic reconstructions of the Greenland and Antarctic ice sheets during the glacial cycles. Quat. Sci. Rev. 2002, 21, 203–231. [Google Scholar] [CrossRef] [Green Version]
- Polyak, V.J.; Onac, B.P.; Fornós, J.J.; Hay, C.; Asmerom, Y.; Dorale, J.A.; Ginés, J.; Tuccimei, P.; Ginés, A. A highly resolved record of relative sea level in the western Mediterranean Sea during the last interglacial period. Nat. Geosci. 2018, 11, 860–864. [Google Scholar] [CrossRef]
- Menviel, L.; Capron, E.; Govin, A.; Dutton, A.; Tarasov, L.; Abe-Ouchi, A.; Drysdale, R.N.; Gibbard, P.L.; Gregoire, L.; He, F.; et al. The penultimate deglaciation: Protocol for Paleoclimate Modelling Intercomparison Project (PMIP) phase 4 transient numerical simulations between 140 and 127 ka, version 1.0. Geosci. Model Dev. 2019, 12, 3649–3685. [Google Scholar] [CrossRef] [Green Version]
- Allouche, O.; Tsoar, A.; Kadmon, R. Assessing the accuracy of species distribution models: Prevalence, kappa and the true skill statistic (TSS). J. Appl. Ecol. 2006, 43, 1223–1232. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org/ (accessed on 20 January 2021).
- Gottlieb, L.D.; Ford, V.S. Phylogenetic relationships among the sections of Clarkia (Onagraceae) inferred from the nucleotide sequences of PgiC. Syst. Bot. 1996, 21, 45–62. [Google Scholar] [CrossRef]
- Ford, V.S.; Gottlieb, L.D. Tribal relationships within Onagraceae inferred from PgiC sequences. Syst. Bot. 2007, 32, 348–356. [Google Scholar] [CrossRef]
- Li, X.; Feng, T.; Randle, C.; Schneeweiss, G.M. Phylogenetic relationships in Orobanchaceae inferred from low-copy nuclear genes: Consolidation of major clades and identification of a novel position of the non-photosynthetic Orobanche clade sister to all other parasitic Orobanchaceae. Front. Plant Sci. 2019, 10, 902. [Google Scholar] [CrossRef] [Green Version]
- López, E.; Devesa, J.A. Estudio taxonómico de Centaurea sect. Acrocentron (Cass.) DC. (Asteraceae) en la Península Ibérica y Baleares. Lagascalia 2013, 33, 75–173. [Google Scholar]
- Gutiérrez Larena, B.; Fuertes Aguilar, J.; Nieto Feliner, G. Glacial-induced altitudinal migrations in Armeria (Plumbaginaceae) inferred from patterns of chloroplast DNA haplotype sharing. Mol. Ecol. 2002, 11, 1965–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrión, J.S.; Fernández, S.; González-Sampériz, P.; Gil-Romera, G.; Badal, E.; Carrión-Marco, Y.; López-Merino, L.; López-Sáez, J.A.; Fierro, E.; Burjachs, F. Expected trends and surprises in the Lateglacial and Holocene vegetation history of the Iberian Peninsula and Balearic Islands. Rev. Palaeobot. Palynol. 2010, 162, 458–475. [Google Scholar] [CrossRef] [Green Version]
- Ben-Menni Schuler, S.; López-Pujol, J.; Blanca, G.; Vilatersana, R.; Garcia-Jacas, N.; Suárez-Santiago, V.N. Influence of the Quaternary glacial cycles and the mountains on the reticulations in the subsection Willkommia of the genus Centaurea. Front. Plant Sci. 2019, 10, 303. [Google Scholar] [CrossRef]
- Vilatersana, R.; Susanna, A.; Garcia-Jacas, N.; Garnatje, T. Karyology, generic delineation and dysploidy in the genera Carduncellus, Carthamus and Phonus (Asteraceae). Bot. J. Linn. Soc. 2000, 134, 425–438. [Google Scholar] [CrossRef]
- Garnatje, T.; Garcia-Jacas, N.; Vilatersana, R. Natural triploidy in Centaurea and Cheirolophus (Asteraceae). Bot. Helv. 2001, 111, 25–29. [Google Scholar]
- Ferriol, M.; Garmendia, A.; Ruiz, J.J.; Merle, H.; Boira, H. Morphological and molecular analysis of natural hybrids between the diploid Centaurea aspera L. and the tetraploid C. seridis L. (Compositae). Plant Biosyst. 2012, 146, 86–100. [Google Scholar] [CrossRef]
- Crawford, D.J.; Smith, E.B. Allozyme divergence and intraspecific variation in Coreopsis grandiflora (Compositae). Syst. Bot. 1984, 9, 219–225. [Google Scholar] [CrossRef]
- Godt, M.J.W.; Hamrick, J.L. Allozyme variation in two Great Smoky mountain endemics: Glyceria nubigena and Rugelia nudicaulis. J. Hered. 1995, 86, 194–198. [Google Scholar] [CrossRef]
- Helenurm, K.; Ganders, F.R. Adaptive radiation and genetic differentiation in Hawaiian Bidens. Evolution 1985, 39, 753–765. [Google Scholar] [CrossRef]
- Ellstrand, N.C.; Roose, M.L. Patterns of genotypic diversity in clonal plant species. Am. J. Bot. 1987, 74, 123–131. [Google Scholar] [CrossRef]
- Segraves, K.A.; Thompson, J.N.; Soltis, P.S.; Soltis, D.E. Multiple origins of polyploidy and the geographic structure of Heuchera grossulariifolia. Mol. Ecol. 1999, 8, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Soltis, D.E.; Soltis, P.S. Polyploidy: Recurrent formation and genome evolution. Trends Ecol. Evol. 1999, 14, 348–352. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E. The role of genetic and genomic attributes in the success of polyploids. Proc. Natl. Acad. Sci. USA 2000, 97, 7051–7057. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subpopulation | N | P95 | A | He | Hi |
|---|---|---|---|---|---|
| Cerro Tudia | 26 | 54.55% | 1.91 | 0.263 | 0.263 |
| El Labrado | 28 | 63.64% | 2.09 | 0.311 | 0.330 |
| Cumbre de los Bonales | 31 | 63.64% | 2.09 | 0.286 | 0.283 |
| Average | – | 60.61% | 2.03 | 0.287 | 0.292 |
| Species | Region | Number of Gaps | Polymorphic Sites | Total Mutations | Parsimony Informative Sites |
|---|---|---|---|---|---|
| C. tentudaica | ETS | 127 | 73 | 75 | 48 |
| AGT1 | 278 | 51 | 51 | 7 | |
| PgiC | 2 | 35 | 37 | 14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreyra, L.D.; Márquez, F.; Susanna, A.; Garcia-Jacas, N.; Vázquez, F.M.; López-Pujol, J. Genesis, Evolution, and Genetic Diversity of the Hexaploid, Narrow Endemic Centaurea tentudaica. Diversity 2021, 13, 72. https://doi.org/10.3390/d13020072
Moreyra LD, Márquez F, Susanna A, Garcia-Jacas N, Vázquez FM, López-Pujol J. Genesis, Evolution, and Genetic Diversity of the Hexaploid, Narrow Endemic Centaurea tentudaica. Diversity. 2021; 13(2):72. https://doi.org/10.3390/d13020072
Chicago/Turabian StyleMoreyra, Lucía D., Francisco Márquez, Alfonso Susanna, Núria Garcia-Jacas, Francisco María Vázquez, and Jordi López-Pujol. 2021. "Genesis, Evolution, and Genetic Diversity of the Hexaploid, Narrow Endemic Centaurea tentudaica" Diversity 13, no. 2: 72. https://doi.org/10.3390/d13020072