
Abstract
Lettuce (Lactuca sativa L.) belongs to the genus Lactuca L. and is an important vegetable worldwide. Over the past decades, there have been many controversies about the phylogeny of Lactuca species due to their complex and diverse morphological characters and insufficient molecular sampling. In this study we provide the most extensive molecular phylogenetic reconstruction of Lactuca, including African wild species, using two chloroplast genes (ndhF and trnL-F). The sampling covers nearly 40 % of the total endemic African Lactuca species and 34 % of the total Lactuca species. DNA sequences from all the subfamilies of Asteraceae in Genebank and those generated from Lactuca herbarium samples were used to establish the affiliation of Lactuca within Asteracaeae. Based on the subfamily tree, we selected 33 ndhF sequences from 30 species and 79 trnL-F sequences from 48 species to infer relationships within the genus Lactuca using randomized axelerated maximum likelihood and Bayesian inference analyses. Biogeographical, chromosomal and morphological character states were reconstructed over the Bayesian tree topology. We conclude that Lactuca contains two distinct phylogenetic clades—the crop clade and the Pterocypsela clade. Other North American, Asian and widespread species either form smaller clades or mix with the Melanoseris species. The newly sampled African endemic species probably should be treated as a new genus.
Similar content being viewed by others


Introduction
Domesticated lettuce (Lactuca sativa L.) is a member of the genus Lactuca L., which is grouped in the subtribe Lactucinae, tribe Cichorieae (Lactuceae), subfamily Cichorioideae of the family Asteraceae (Compositae; Judd et al. 2007; Kadereit et al. 2007). As one of the most important vegetables, lettuce is commercially produced worldwide, especially in Asia, North and Central America, and Europe (Lebeda et al. 2007). There are a large number of lettuce cultivars within L. sativa. These cultivars can be divided in seven distinct cultivar groups: Butterhead Group, Crisphead Group, Cos Group, Cutting Group, Stalk Group, Latin Group and Oilseed Group (de Vries 1997). Many studies have focused on domesticated lettuce (Hartman et al. 2012; Kerbiriou et al. 2013; Uwimana et al. 2012; Zhang et al. 2009a, b). However, there are still uncertainties about the phylogenetic relationships within Lactuca, mainly due to the complex and variable morphological characters of the species in the genus. Some of the controversies stem from the different circumscriptions proposed for the genus, which vary from extremely broad to very narrow concepts. Bentham (1873) included Lactuca species not only from the present subtribe Lactucinae, but also from the present subtribes Crepidinae and Hyoseridinae; this broad concept was maintained by Hoffmann (1890–1894). Stebbins (1937a, b, 1939), Feráková and Májovský (1977) and Lebeda et al. (2004, 2007) used a moderately wide concept of Lactuca that comprised a total of approximately 100 species. Tuisl (1968), Shih (1988a, b), and Kadereit et al. (2007) established a narrow circumscription. In this concept, Shih and Kilian (2011) consider there to be between 50 and 70 Lactuca species. However, all these authors mentioned before only dealt mostly with regional Lactuca species and the genus has never been revised in its entirety.
Lebeda et al. (2004) provided an overview of the biogeographical distribution of wild Lactuca species based on the available literature data and showed that Asia (containing 51 species) and Africa (containing 43 species) are the two centres of diversity for Lactuca species. Lebeda et al. (2004, 2009) elaborated a classification of Lactuca from taxonomic and biogeographical criteria and divided the genus into seven sections (Lactuca (subsection Lactuca and Cyanicae DC.), Phaenixopus (Cass.) Bentham, Mulgedium (Cass.) C.B. Clarke, Lactucopsis (Schultz Bip. ex Vis. et Pančić) Rouy, Tuberosae Boiss., Micranthae Boiss., Sororiae Franchet) and two geographical groups (African and North American). Recently, Wang et al. (2013) constructed a DNA-based phylogenetic tree of the Lactuca alliance with a focus on the Chinese centre of diversity. This study fills the gap in our understanding of Asian diversity centre of Lactuca species and related genera, especially for the Chinese species. However, a study of the African diversity centre of Lactuca species is still lacking.
Despite the lack of studies focused on the entire Lactuca genus, there have been a number of studies focused on cultivated lettuce and closely-related wild species. These studies concentrated on aspects of interest for lettuce breeding to improve growth related to abiotic and biotic stresses using genetic resources from wild lettuce species (Hartman et al. 2012, 2014; Jeuken et al. 2008; van Treuren et al. 2011). Zohary (1991) established a concept of the ‘lettuce gene pool’ and Koopman et al. (1998, 2001) modified Zohary’s lettuce gene pool concept and provided the first molecular phylogenetic relationships among Lactuca species based on nrDNA ITS-1 and AFLPs. Koopman et al. (1998) described L. sativa, L. serriola L., L. dregeana DC., L. aculeata Boiss. and L. altaica Fischer et C.A. Meyer as the primary gene pool, L. virosa L. and L. saligna L. as the secondary gene pool, and L. quercina L., L. viminea, L. sibirica Benth. ex Maxim. and L. tatarica (L.) C.A. Meyer as the tertiary gene pool. Apart from Koopman et al. (2001) and Wang et al. (2013), there is limited information about the molecular phylogenetic relationships within the genus Lactuca, especially for the African species since they were first described (Jeffrey 1966; Stebbins 1937b).
More than 4000 years ago, the Egyptians started to cultivate wild lettuce (L. serriola) in Africa and this species is thought to be the ancestor of modern lettuce cultivars (Harlan 1986). Lindqvist (1960) doubted that only L. serriola was involved in the domestication of the cultivated lettuce, but he did not specify what species might have played a role. Kesseli et al. (1991) suggested a polyphyletic origin of L. sativa using RFLP loci. Mikel (2007) reported that apart from L. serriola, the current crisphead cultivar ‘Salinas’ was also derived from L. virosa for its robust root system and decreased leaf drop. Wei et al. (2014), using a recombinant inbred line population derived from L. sativa ‘Salinas’ (crop) and L. serriola (wild), found that alleles from the cultivated lettuce contribute more to lateral root development than those from wild lettuce.
The aim of this present study is to provide a DNA based phylogenetic tree of Lactuca, and 34 % of known Lactuca species and 40 % of the total endemic African Lactuca species were included in the taxon sampling. We reconstruct ancestral states for geographic areas, chromosome number and selected morphological characters over the phylogenetic trees. Novel potential genetic resources for lettuce breeding are proposed as well.
Materials and methods
Taxon sampling
Twenty-seven Lactuca species, including thirteen African endemic species, and four species from Lactuca-allied genera were sampled (Table 1). For the species L. viminea two samples representing two subspecies were included. Following the treatment of Lebeda et al. (2004), this sampling represents 34 % of the total Lactuca species and 40 % of the total endemic African species. The 32 samples come from fresh leaf, sillica-dried leaf and herbarium specimens (Table 1). Four of the fresh-collected materials were from Centre for Genetic Resources, the Netherlands (CGN, http://www.wageningenur.nl/en/Expertise-Services/Statutory-research-tasks/Centre-for-Genetic-Resources-the-Netherlands-1.htm). Herbarium materials were provided by the National Herbarium of the Netherlands (WAG) and the Botanic Garden and Botanical Museum Berlin-Dahlem (B), herbarium codes following Thiers (2011). All necessary permissions for the described plants and specimen samplings were obtained from the respective curators, dr. ir. J.J. Wieringa (Naturalis Biodiversity Center, Leiden) and dr. Norbert Kilian (Botanic Garden and Botanical Museum Berlin-Dahlem, Freie Universität Berlin, Berlin).
DNA extraction and purification
DNA was extracted from 10 to 30 mg of plant material using the cetyltrimethyl-ammonium-bromide (CTAB) method (Doyle and Doyle 1987), modified for herbarium specimens as in Särkinen et al. (2012) and Staats et al. (2011). The DNA extraction was then purified by Wizard DNA clean-up system (Promega Corp.) with a vacuum manifold (Promega Corp.) The quality of the DNA extractions was visualized on 1 % agarose gel and measured by Qubit 2.0 Fluorometer (Invitrogen). Polymerase chain reaction and Sanger sequencing were also performed for some of the herbarium samples to check for potential degradation of DNA. PCR amplifications were performed in 10 μl reactions using MyTaq™ DNA polymerase (Bioline, London, UK). Thermal cycling for PCR included 2 min at 95 °C, followed by 30 cycles of 30 s at 95 °C, 30 s at 50 °C, 1 min at 70 °C, and ended by 5 min at 72 °C. The forward and reverse primer sequences of trnL-F were 5′-GCAATCCTGAGCCAAATCC-3′ and 5′-GCTCGATGCATCATCCCGCTAAA-3′, respectively. Two pairs of primers (ndhF 5′ forward-1074 reverse and 913 forward-ndhF 3′ reverse) were used for the amplification of ndhF due to the large size of the gene (Karis et al. 2001). PCR products were then purified and sequenced as described in Schneider et al. (2014).
Next generation sequencing and de novo assembly
The dataset of plastid gene sequences presented in this work was generated as part of the SYNTHESYS Joint Research Activities 4 (JRA4: Plants/fungi herbarium DNA: http://www.synthesys.info/joint-research-activities/synthesys-2-jras/jra4-plantsfungi-optimised-dna-extraction-techniques/). The Lactuca samples were sequenced by National High-Throughput DNA Sequencing Centre of University of Copenhagen, using the next generation sequencing Illumina HiSeq 2000 platform (http://seqcenter.ku.dk/facilities/). The protocols for DNA library preparation and PCR amplification was described in Bakker et al. (2015). Contig assembly and read clean-up were performed using standard method similar to the ‘MitoBIM’ approach outlined in Hahn et al. (2013) for mitochondrial genomes. This method is called the Iterative Organelle Genome Assembly pipeline (IOGA), aiming to assemble paired-end reads into a series of candidate assemblies and selecting the best one based on likelihood estimation (Bakker et al. 2015). The IOGA pipeline can be briefly described in the following steps: (1) Trimmomatic was used to trim low quality, adapter and other Illumina-specific sequences from individual reads (Bolger et al. 2014); (2) chloroplast genome-derived reads were filtered out of the entire read pool in Bowtie 2, by aligning the latter to a range of reference Angiosperm chloroplast genome sequences (Langmead and Salzberg 2012); (3) de novo assemblies from the trimmed, filtered and corrected chloroplast reads, were performed in SOAPdenovo2, using k-mer values ranging from 37 to 97 (Luo et al. 2012); (4) ‘best assemblies’ were selected using the N50 criterion and then used as a new reference to find target-specific reads not selected in the first iteration; (5) step 4 was repeated until no more chloroplast genome-derived reads were found, followed by assembly of the final set of assemblies with SPAdes3.0 (Bankevich et al. 2012), under a range of different k-mer settings; (6) finally, Assembly Likelihood Estimation (Clark et al. 2013) was performed to select the best assembly (LnL score) among candidate assemblies as the final assembly. Chloroplast genes (trnL-F and ndhF) were annotated and extracted in DOGMA (Wyman et al. 2004). The IOGA script can be obtained from Github at https://github.com/holmrenser/IOGA.
Sequence alignment and phylogenetic analyses
From GenBank we obtained 218 ndhF gene sequences from 211 species and 301 trnL-F gene sequences from 250 species by Blasting L. sativa, L. inermis Forssk., L. paradoxa Sch.Bip. ex A. Rich. and L. canadensis A. Gray (Table S1 and Table S2) against the NCBI nucleotide database. This sampling comprises a wide range of taxa from all the subfamilies in Asteraceae, according to the Angiosperm Phylogeny Website (http://www.mobot.org/MOBOT/research/APweb/). Together from with the Lactuca sequences generated in this study, we achieved 34 % taxonomic sampling for Lactuca. Barnadesia caryophylla was selected as outgroup based on the phylogenetic tree of Asteraceae in APG (http://www.mobot.org/MOBOT/research/APweb/trees/asteraceae.gif). All the DNA sequences were first automatically aligned with MAFFT (version 7, http://mafft.cbrc.jp/alignment/server/; Katoh et al. 2002) and then manually adjusted in Mesquite 2.75 (Maddison and Maddison 2015), following the criteria used by Borsch et al. (2003), Bremer et al. (2002), Kim and Jansen (1995) and Taberlet et al. (2007). The alignments for trnL-F and ndhF genes were separately optimised by first performing Neighbour Joining in PAUP* version 4.0b10 (Swofford 2003). The following parameters were used: Outgroup: Barnadesia caryophylla, Dset Distance = GTR, Rates = Gamma. The vertical order of accessions in the two alignments was then adjusted according to the NJ tree in order to maintain a phylogenetic continuum and to see if local rearrangements in the alignment of nucleotides were needed. Presumably homologous indel events (gaps) were coded as additional presence/absence characters. Regions left doubts about the homology of indels or could not be aligned were treated as in Bremer et al. (2002).
Phylogenetic trees at the subfamily level were then reconstructed for ndhF and trnL-F regions separately using Randomized Axelerated Maximum Likelihood (RAxML)-HPC2 run on XSEDE (Stamatakis 2014) from the Cyber-infrastructure for Phylogenetic Research (CIPRES) Science Gateway (V. 3.3, available at http://www.phylo.org/; Miller et al. 2010; Figure S1 & S2). Simultaneously, MrBayes 3.2.2 on XSEDE from CIPRES Science Gateway was also used to perform phylogenetic analyses (Ronquist et al. 2012), using the same alignment (Figure S3 & S4).
In order to estimate phylogenetic relationships at the generic level, we then subsampled our subfamily level alignments based on the generated trees (Fig. S1–S4) and trees from Wang et al. (2013). 79 trnL-F and 33 ndhF accessions were selected to represent Lactuca and related genera. Leontodon saxatilis is the nearest sister group to Lactuca and related genera and therefore was chosen as the outgroup (Fig. S1 - S4). The subsampled sequences were re-aligned using MAFFT version 7. Indels were manually coded for trnL-F and ndhF genes following the Simple Indel Coding (SIC) method (Simmons and Ochoterena 2000) in Mesquite 2.75. The selected sequences were then concatenated using SequenceMatrix-Windows 1.7.8 (Vaidya et al. 2011).
The joined alignment, containing the two plastid DNA sequences, as well as the two separate gene alignments were used for further phylogenetic analyses. For the joined alignment, the dataset was analysed in three different ways for Bayesian Inference (BI): no partition, two partitions (trnL-F/ndhF) and three partitions (trnL-F/codon position1 + 2 of ndhF/codon position 3 of ndhF). The parameters for BI were as follows: outgroup Leontodon saxatilis; lset nst = mixed, rates = gamma; unlink statefreq = (all), revmat = (all), shape = (all), pinvar = (all); prset applyto = (all), ratepr = variable; mcmcp ngen = 50,000,000, relburnin = yes, burninfrac = 0.25, printfreq = 1000, samplefreq = 50,000 nchains = 4 temp = 0.05; Report tree = brlens. Other parameters were default settings. For the single gene alignments, the dataset of ndhF gene was treated in two ways for BI: no partition and two partitions (codon position1 + 2/codon position 3) and the alignment of trnL-F gene was not partitioned as it is not a coding sequence.
The Markov Chain output parameter files generated by MrBayes 3.2.2 were then used in Tracer v1.6 (available at http://tree.bio.ed.ac.uk/software/tracer/) to select the best partition for constructing phylogenetic trees by selecting the marginal density centred around the highest log likelihood (LnL). The chosen partition was then subjected to RAxML analysis using default settings. TreeGraph 2 was used to add Bootstrap (BS) and Posterior Probability (PP) values on one tree (Stover and Muller 2010).
Biogeographical, chromosomal and morphological data analyses
Biogeographical distributions were inferred from The Cichorieae Portal (Hand et al. 2009+) and Lebeda et al. (2004). We used RASP (Reconstruct Ancestral State in Phylogenies) to reconstruct ancestral biogeographical areas whereby distribution areas were delineated as A(Asia), B(Europe), C(Africa) and D(North America) (Yu et al. 2015). We did not delineate more detailed distributions due to the restriction of the number of biogeographical areas in RASP. We used 1000 trees inferred from BI analyses and the condensed Bayesian tree in RASP. The Bayesian Binary MCMC (BBM; Experimental) method and the Fixed (JC) + Gamma model were used to reconstruct the biogeographical areas. Other settings were default.
Chromosome numbers were scored according to Koopman et al. (1993), Matoba et al. (2007) and the Index to Plant Chromosome Numbers (IPCN; Missouri Botanical Garden 2014). Selected morphological characters, such as floret number, achene winged or not and rib number were scored from The Cichorieae Portal (Hand et al. 2009+). We selected these characters because they are considered as important identification keys. Subsequently, we reconstructed the ancestral states for chromosomal and morphological characters over the same trees used for estimating the ancestral state of the biogeographical data in RASP. All the settings were the same.
Results
The ndhF and trnL-F sequences of 27 species were successfully sequenced by NGS, whereas the sequences of L. praevia C.D. Adams and L. viminea J. Presl & C. Presl subsp. ramosissima (All.) Malag. were failed for NGS and obtained using Sanger sequencing. In addition, the sequencing of L. imbricata Hiern, L. orientalis Boiss. and L. schulzeana Büttner was neither successful by NGS or Sanger. The trnL-F region had 863 (including indels)/853 characters in the alignment. Of the total 863/853 characters, 65(7.5 %)/58(6.8 %) were parsimony informative sites (Table 2). The alignment of ndhF gene contained 2251 (including indels)/2250 characters and 71(3.2 %)/70(3.1 %) of them were informative sites (Table 2). The total number of characters in the concatenated alignment was the sum of trnL-F and ndhF and 136(4.4 %)/128(4.1 %) of them were informative sites. The phylogenetic trees of 247 ndhF and 331 trnL-F gene sequences from different subfamilies using RAxML and BI analyses are shown in Fig. S1–S4. The no partition model for the concatenated dataset performed better than the partition models, as its marginal density was centred around a higher log likelihood (LnL), and therefore was chosen for further analyses. One ‘best ML tree’ for the concatenated sequences was inferred automatically from the RAxML analysis, which is generally congruent in topology with the BI 50 % majority rule consensus tree. We present the RAxML phylogram topology combined with BS and PP values (Fig. 1). The phylogenetic trees for single gene alignments are shown in Figs. S5 and S6. We also reconstructed ancestral states for biogeographical, chromosomal and morphological characters over the condensed Bayesian trees of the concatenated sequences (Figs. S7–S11).

RAxML phylogram (‘best ML tree’) of the concatenated sequences of ndhF gene and trnL-F gene used in this study; Bootstrap (BS > 50) support values are given above the branches and Posterior Probability (PP > 0.5) support values are below; the names of Chinese taxa are referred to Wang et al. (2013); star L. tinctociliata was mis-identified and it could be Launaea cornuta; L. ugandensis should be Lactuca sp.
The phylogenetic analyses showed that L. tinctociliata I.M. Johnst is outside the Lactuca clade and the sister group to all Lactuca and Melanoseris species, Notoseris triflora (Hemsl.) C. Shih, Paraprenanthes diversifolia (Vaniot) N. Kilian, Cicerbita alpina Wallr. and Prenanthes purpurea (Vaniot) N. Kilian (Fig. 1, name indicated with a star). A Lactuca clade (BS = 78, PP = 0.98) divides into three clades, Clade A, B and C. We will describe the clades in the following sections.
Clade 1 (BS = 95, PP = 1) includes the lettuce crop and closely related wild lettuce species. It contains two subclades. Clade 1a (BS = 97, PP = 0.99) consists of the domesticated lettuce L. sativa and its closest relatives L. serriola, L. altaica, L. aculeata, L. saligna and L. virosa. One L. serriola accession is the sister group to L. altaica (BS = 66, PP = 0.76). L. aculeata and L. sativa are grouped together (BS = 63, PP = 0.98). L. saligna and L. virosa are the sister groups of L. serriola, L. altaica, L. aculeata and L. sativa. Clade 1b (BS = 100, PP = 1) comprises L. orientalis, L. viminea J. Presl et C. Presl, L. viminea J. Presl et C. Presl subsp. chondrilliflora (Boreau) Malag. and L. viminea subsp. ramosissima. Clade 1 (PP = 1) comprises widely spread Lactuca species from Asia, Europe and Africa (Figure S7). The species in Clade 1 have a chromosome number of eighteen (2n = 18) except L. orientalis (2n = 18 or 36; Figure S8). Most species in Clade 1a have a floret number between 6 and 15 (20) or even more than 20 florets (Figure S9). Other species in Clade 1b have less than 6 florets (Figure S9). The achenes of most species in Clade 1 are not winged except L. virosa (Figure S10). Most species in Clade 1 have a rib number between 3 and 9 (Figure S11).
Clade 2 (BS = 99, PP = 1) comprises of ex-Pterocypsela C. Shih species, including L. indica L., L. raddeana Maximowicz, L. formosana Maximowicz and L. ugandensis C. Jeffrey (not ex-Pterocypsela species). Four L. indica accessions, one L. raddeana accession and L. ugandensis are in one subclade (BS = 89, PP = 1) whereas the other three L. raddeana accessions and four L. formosana accessions are in one clade (BS = 50). In addition, one L. tatarica accession is the sister group to Clade 2, though the BS support is very low (BS < 50). This clade contains Asian species and one African species L. ugandensis clade (PP = 1; Figure S7). Lactuca species in Clade 2 have eighteen chromosomes (2n = 18) but this information for L. ugandensis is missing (Figure S8). They usually have a floret number between 6 and 15 (sometimes more than 20; Figure S9). Most species in Clade 2 (excluding L. ugandensis) have winged achenes (Figure S10) and a rib number between 1 and 7 (Figure S11).
Clade 3 (BS = 82, PP = 1) consists of L. dolichophylla Kitamura, L. dissecta D. Don and L. tuberosa Jacq. Clade 4 (lacking support) is composed of L. tenerrima Pourr., L. inermis and L. canadensis. L. inermis 1 from Ghana is the sister group of L. tenerrima, L. canadensis and L. inermis 2 from Togo. Clade 5 (BS = 100, PP = 1) includes L. undulata Ledebour and L. perennis L. Clade 6 (BS = 96, PP = 1) contains two L. tatarica accessions and L. sibirica. Clade 3 and 4 (PP = 1) include species from Asia and widespread species (Figure S7). Most species in Clade 5 and 6 are from Asia, North America or widespread species (Figure S7). The Lactuca species in Clade 3 have sixteen chromosomes (2n = 16; Figure S8). Lactuca species in Clade 5 and 6 have a chromosome number of eighteen (2n = 18). L. tenerrima and L. inermis in Clade 4 have sixteen chromosomes (2n = 16) while L. canadensis has thirty-four chromosomes (2n = 34; Figure S8). Most species in Clade 3–6 have a floret number usually between 6 and 15 (sometimes more than 20; Figure S9) and non-winged achenes (excluding L. canadensis and L. tuberosa (Figure S10). Most species in Clade 3 and 4 have a rib number between 3 (1) and 7. Species in Clade 5 and 6 have 1–3 ribs (Figure S11).
Clade 7 contains four Parasyncalathium souliei (Franch.) J.W. Zhang, Boufford et H. Sun accessions with a good support value (BS = 99, PP = 1; Fig. 1). Clade 8 lacks support (BS < 50, PP = 0.69) but may become stronger after adding more taxonomic sampling. It includes Melanoseris cyanea Edgew, M. violifolia (Decne.) N. Kilian, M. atropurpurea (Franch.) N. Kilian et Ze H. Wang and M. macrantha (C.B. Clarke) N. Kilian et J.W. Zhang. Other Melanoseris species, M. atropurpurea, M. qinghaica (S.W. Liu et T.N. Ho) N. Kilian et Ze H. Wang, M. macrorhiza (Royle) N. Kilian, M. likiangensis (Franch.) N. Kilian et Ze H. Wang are in a huge polytomy. Melanoseris and Parasyncalathium species are from Asia or widespread species (Figure S7). They have sixteen chromosomes (2n = 16; Figure S8). Melanoseris species have a floret number between 6 and 15 (sometimes more than 20) while Parasyncalathium souliei has a floret number less than 6 (Figure S9). Melanoseris and Parasyncalathium species do not have winged achenes (Figure S10). The rib number of most Melanoseris species is unknown (Figure S11). Parasyncalathium souliei in Clade 8 has 1–3 ribs.
Clade B (BS = 99, PP = 1) contains three scandent African species, L. glandulifera Hook.f., L. attenuata Stebbins and their sister group L. paradoxa (Figure S7). Clade C (PP = 0.58) includes the African species L. lasiorhiza (O. Hoffm.) C. Jeffrey, L. schweinfurthii Oliv. et Hiern, L. calophylla C. Jeffrey, L. zambeziaca C. Jeffrey, L. setosa Stebbins ex C. Jeffrey, L. praevia and Melanoseris bracteata (Hook.f. et Thomson ex C.B. Clarke) N. Kilian. Chromosome number is only available for L. attenuata (2n = 32) and L. glandulifera (2n = 16; Figure S8). Species in Clade B and C have a floret number less than 6 (Figure S9) and they do not have winged achenes (Figure S10). Most species in Clade B have a rib number between 3 and 7. Species in Clade C have 1–3 ribs (Figure S11).
Discussion
Lettuce is an economically important crop and consequently most studies have mainly focused on L. sativa and closely related wild species (Koopman et al. 1993, 1998, 2001). Conversely, the entire Lactuca genus is poorly studied, especially for the two regions with the highest diversity, Asia (51 species) and Africa (43 species; Lebeda et al. 2004). Recently, a publication focused on the Chinese centre of diversity, including 15 Asian Lactuca species (Wang et al. 2013). However, the African Lactuca center of diversity remains unstudied. We here present the first study focused on the phylogenetic relationships within Lactuca and related genera with extensive sampling of the African diversity centre, based on plastid genes. This is the first molecular phylogeny for 40 % of the endemic African Lactuca species, especially for the scandent species since they were described and revised by Stebbins (1937b).
The mapping of biogeographical, chromosomal and morphological character states lend additional supports to the topologies of the RAxML trees. For biogeographical data, Clade B and Clade C only contain Lactuca species endemic to African continent, although other clades do not show distinctive pattern. The chromosome numbers (excluding the accessions with unknown chromosome number in Clade 8) supported the topology of the RAxML tree. Lactuca species in Clade 1, 2, 5 and 6 have a chromosome number of eighteen (2n = 18) except L. orientalis (2n = 18 or 36). Species in Clade 3, and Melanoseris species have sixteen chromosomes (2n = 16). L. tenerrima and L. inermis in Clade 4 have sixteen chromosomes (2n = 16) while L. canadensis has thirty-four chromosomes (2n = 34). In Clade B, L. glandulifera has sixteen chromosomes (2n = 16) while L. attenuata has thirty-two (2n = 32). The floret number also validated the topology of the RAxML tree. Most species in Clade 1a, 2–6 and C have a floret number usually between 6 and 15 (sometimes more than 20). Other species in Clade 1b, 7, B and C have a floret number less than 6. For the state of achene, most species in the Lactuca clade do not have winged achenes. Only L. virosa, L. canadensis, L. tuberosa and species in Clade 2 (excluding L. ugandensis) have winged achenes. For rib number, most species in Clade 1, 4 and B have a rib number between 3 and 9. Species in Clade C, 5, 6 and Clade 8 have 1–3 ribs. Species in Clade 2 and 3 have a rib number between 1 and 7. The rib number of most Melanoseris species is unknown.
Monophyly of the subtribe Lactucinae
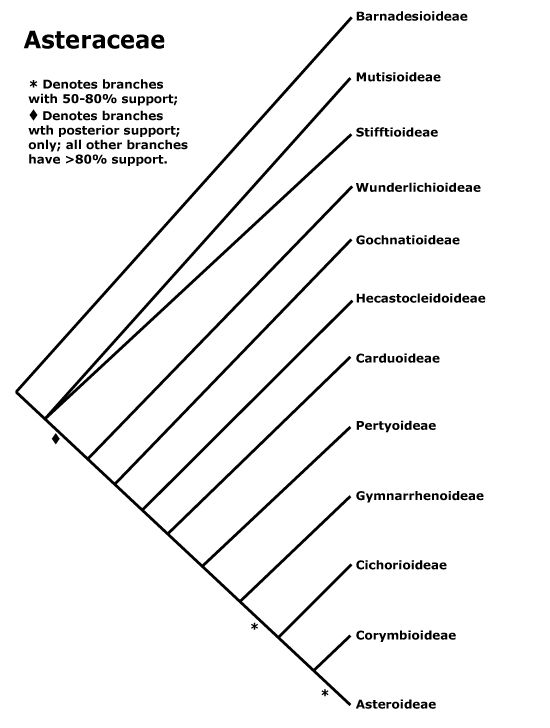
Our RAxML tree for concatenated sequences shows that C. alpina, Faberia, P. purpurea and L. tinctociliata should be excluded to maintain the monophyly of the subtribe Lactucinae (Figs. S1–S4). L. tinctociliata is placed outside Lactucinae and nested in Hyoseridinae (Figs. S1–S4). It is clustered with Launaea sarmentosa (Willd.) Kuntze with a very high support (BS = 100, PP = 1) in the trnL-F tree and is sister group of Sonchus oleraceus L. in the ndhF trees (BS < 50, PP = 0.64; Figs. S1–S4). This species was first published and described by I.M. Johnst in 1925 (Jeffrey 1966; Anonymous 1925). No detailed description or molecular data have been made available since then. According to I.M. Johnst, L. tinctociliata is very well characterized by its narrow firm purple leaf-margins which commonly bear purplish-tinged teeth and fleshy cilia, the capitula with about 12 yellow flowers, a very compressed achene, marginal, oblong-ovate or oblanceolate 5–6 mm long, thin beak >1 mm long, about 12 ribs, bristle white pappus, 5–6 mm long (Anonymous 1925). From the image of the L. tinctociliata specimen used in this study, we can see (image available at http://medialib.naturalis.nl/file/id/WAG.1288514/format/large?width=800px&height=800px) that it has broader leaves than the type specimen (image available at http://plants.jstor.org/stable/10.5555/al.ap.specimen.gh00009514) and does not have purple leaf-margins. Although we could only compare the specimen images, the ‘L. tinctociliata’ used in our study is clearly not L. tinctociliata. Based on our molecular data and the woody habit (typical of the species), the specimen is most likely Launaea cornuta (Hochst. ex Oliv. et Hiern) C. Jeffrey.
Wang et al. (2013) indicated that when Faberia and P. purpurea lineages are excluded, the subtribe Lactucinae is monophyletic. Moreover, they suggested that C. alpina should be disregarded while the other Cicerbita species are placed inside the Lactucinae. A narrow circumscription of Prenanthes L. was proposed making it a probably monospecific genus (Kilian and Gemeinholzer 2007; Kilian et al. 2009). Wang et al. (2013) transferred species from Prenanthes to Notoseris Shih and confirmed this narrow concept of Prenanthes. The BI tree of ndhF, including species from different subfamilies (Figure S3), shows that the genus Tolpis Adanson from the subtribe Cichoriinae is the sister group of the clade comprising P. purpurea, C. alpina, N. triflora, Paraprenanthes diversifloria and the genus Lactuca (PP = 0.54), but support for this pattern is lacking. The RAxML ndhF tree indicates P. purpurea is the sister group of Tolpis species (Figure S1). In our trnL-F trees, P. purpurea is the sister group of Ixeridium gracile (DC.) C. Shih, a species from the subtribe Crepidinae (BS = 61, PP = 0.93; Figs. S2, S4). Although all BS and PP values involved are low, these results would confirm the narrow concept of Prenanthes and indicate that P. purpurea probably belongs to the subtribe Cichoriinae or Crepidinae and is far away from the subtribe Lactucinae.
Our RAxML tree reveals that Notoseris and Paraprenanthes C. C Chang ex C. Shih are the sister groups to Lactuca in the subtribe Lactucinae (Fig. 1). When the genus Notoseris was first described, it comprised 12 species, with shared morphological characters such as capitula with 3–5 florets, beakless achene apices and 6–9 ribs on each side of achene (Shih 1987). Shih (1997) then reduced the number of species to 11. Wang et al. (2013) recently removed several species from Notoseris and transferred two scandent species from Prenanthes to Notoseris, based on ITS and plastid DNA sequences. Paraprenanthes was first proposed by C. C. Chang and formally established by Shih (1988a), who added new species and transferred some species from Lactuca, Crepis L. and Mycelis Cass. based on morphological characters, e.g. capitula with 6–23 cyanic florets, achenes with 5 main ribs and two rather similar secondary ribs in-between, and a single pappus (1988a). Shih and Kilian (2011) maintained the circumscription of Paraprenanthes but used a wider species concept and separated three species from the genus. Recently, Wang et al. (2013) revised the genus by reducing the species recognized by Shih and Kilian (2011) to six and adding four new species. Although the phylogenetic relationships among Paraprenanthes and Notoserisspecies remains unresolved based on trnL-F DNA sequence comparisons (Figs. S2, S4), our results indicate that Notoseris and Paraprenanthes are closely related to Lactuca.
Circumscription of Lactuca and its subgeneric classification
The phylogenetic tree for the concatenated sequences indicates that the Lactuca species, autochthonous to the African continent, are far away from the other Lactuca species. Meanwhile, the other Lactuca species (not endemic to Africa), Melanoseris and Paracyncalathium are nested within Clade A (lacking support) as part of the large polytomy (Fig. 1).
The African Lactuca species (Clade B and C, 2n = 16, 32 or ?) The African species include L. paradoxa, L. attenuata, L. glandulifera, L. lasiorhiza, L. schweinfurthii, L. calophylla, L. zambeziaca, L. setosa and L. praevia. Of all of these species we present, as far as we know, the first molecular phylogeny since they were summarized and described by Jeffrey (1966). Jeffrey (1966) elaborated a total of 33 African Lactuca species but Lebeda et al. (2004) reported that this group contains at least 43 species and 75 % of the group (31 in total) can be considered as endemic. In our sampling, only autochthonous African Lactuca species are included in these two clades with one exception—M. bracteata. The support between L. praevia and M. bracteata is very low), hence it is difficult to tell if M. bracteata does or does not belong to Clade C. Other species occuring in Africa but not endemic to the African continent, such as L. inermis, L. tenerrima, L. saligna and L. virosa, are distributed in other clades. This may indicate an independent evolution of the African endemic species. Based on their scandent or herbal habits, these endemic species can be divided into two groups: the scandent group and the herbal group. According to Stebbins (1937b), there were seven scandent Lactuca species in Africa: L. stipulata Stebbins, L. elgonensis Stebbins, L. paradoxa, L. attenuata, L. semibarbata Stebbins, L. wildemaniana Stebbins, and L. glandulifera. Jeffrey (1966) combined the last two species as L. glandulifera and added L. attenuatissima Robyns to the scandent group. Our scandent samples include L. paradoxa, L. attenuata and L. glandulifera. These scandent species are not related to the two scandent species from Notoseris, which indicates two independent evolutions of the scandent habit in Lactucinae (Figs. S2, S4). These African species share some characters, such as capitula with less than 6 yellow florets (an exception from L. lasiorhiza with 10–14 florets) and 1–3 ribs on each side of achene. Chromosome number is only available for L. attenuata (2n = 32) and L. glandulifera (2n = 16; Missouri Botanical Garden 2014). Wang et al. (2013) used the same dataset of Melanoseris species as in our study and showed that the genus Melanoseris is closely related to the genus Lactuca. In our results, Melanoseris and Parasyncalathium species are in Clade A and the African Lactuca species in Clade B and C are even further away from other Lactuca species in Clade A than Melanoseris and Parasyncalathium species. Our molecular, biogeographical, chromosomal and morphological data all show that the endemic African Lactuca species have a unique position and evolved independently. We suggest that the African species in Clade B and Clade C could be removed from Lactuca and treated as a new genus. However, further taxonomic, cytological and molecular studies are still needed to do an official taxonomic revision.
The Melanoseris species (Clade 7 and 8, 2n = 16 or ?) Clade 7 contains Parasyncalathium souliei accessions with a very high support value (BS = 99, PP = 1; Fig. 1). This implication is in line with Stebbins (1940) and Zhang et al. (2009a, b, 2011). However, Wang et al. (2013) preferred to put this species in Melanoseris while Zhang et al. (2011) proposed that this species should be either put back in Lactuca or treated as a new genus. Clade 8 includes M. cyanea, M. violifolia, M. atropurpurea and M. macrantha. One M. atropurpurea accession is in this clade while other three M. atropurpurea accessions are in an unresolved polytomy together with M. macrorhiza, M. likiangensis and M. qinghaica. The name Melanoseris was first proposed by Decaisne in 1843 for two species from the Himalayas, which are now treated as M. lessertiana. Edgeworth (1846) then added more Himalayan species to Melanoseris. Shih (1991) established two new genera from Sino-Himalayan region, Chaetoseris C. Shih and Stenoseris C. Shih, by transferring species from Lactuca and Cicerbita. Chaetoseris was distinguished from Lactuca and Cicerbita because of its achene corpus with broad and thickened lateral ribs and a pappus with an outer ring of minute hairs (Shih 1991, 1997). Stenoseris was established with five species and circumscribed by 3–5 flowered capitula and an achene with an outer ring of minute hairs (Shih 1991). Shih and Kilian (2011) revised this lineage and reused the name Melanoseris for the lineage based on their molecular data. They transferred species that were formerly placed in Chaetoseris, Cicerbita, Lactuca, Mulgedium Cass., Prenanthes and the genus Stenoseris to Melanoseris. Furthermore, Wang et al. (2013), using nrITS1 and plastid genes, concluded that Melanoseris could be divided into three groups: M. cyanea group, M. macrorhiza group and M. graciliflora group. Although our results do not separate the Melanoseris lineage from Lactuca species, they reveal a close relationship between Lactuca and Melanoseris. Compared with previous molecular and morphological investigations, we still think Melanoseris and Lactuca are two separate but closely related genera (Shih and Kilian 2011; Wang et al. 2013).
We will now discuss the clades (1–6) that can be highlighted within Lactuca:
Clade 1 (The Crop Clade) (2n = 18 or 36) This clade comprises Clade 1a and 1b. Clade 1a contains the cultivated lettuce and can be referred to as Lactuca section Lactuca subsect. Lactuca (Lebeda et al. 2009). This clade includes L. serriola, L. altaica, L. aculeata, L. virosa and L. saligna. All the species in Clade 1a are interfertile or partly interfertile with L. sativa (Hartman et al. 2012; Thompson et al. 1941). Koopman et al. (1998) considered L. serriola and L. altaica to be conspecific based on their identical ITS-1 sequences and the results of crossing experiments. Our phylogenetic tree confirms his conclusion and also show that L. aculeata is closer to L. sativa than L. serriola. L. sativa, L. serriola, L. altaica and L. aculeata comprise the primary lettuce gene pool (Koopman et al. 1998). L. virosa and L. saligna are the sister groups to the species in the primary gene pool and form the secondary lettuce gene pool (Koopman et al. 1998). Crosses between L. serriola and L. saligna, and between L. sativa and L. saligna were shown to be partly fertile or self-fertile (Jeuken et al. 2001; Thompson et al. 1941; Zohary 1991). Chromosomal studies have demonstrated that L. saligna is potentially more closely related to L. sativa—L. serriola than L. virosa (Koopman et al. 1993; Matoba et al. 2007). Conversely, nrITS1 and AFLP fingerprints with moderate support indicated that L. virosa is closely-related to L. sativa—L. serriola (Koopman et al. 1998, 2001). Although the cross between L. virosa and L. sativa often failed, it was still possible to obtain the cross and the hybrid was found to be self-sertile (Thompson et al. 1941; Whitaker and Thompson 1941; Zohary 1991). All the species in Clade 1a are widespread and share some characters, like a floret number >6 (Figs. S7–S11).
Clade 1b includes L. orientalis and L. viminea and refers to section Phaenixopus (Lebeda et al. 2009). L. orientalis and L. viminea belonged to the genus Scariola but recently they were both treated as Lactuca species (Flann et al. 2010; Shih 1997; Shih and Kilian 2011; Wang et al. 2013). L. orientalis (2n = 18, 36) is a subshrub, which is very rare in Lactuca, all the other Lactuca species are herbs (Shih and Kilian 2011). It has whitish, rigid, intricately and divaricately branched stems, glaucous green leaves, solitary capitula with 4 or 5 pale yellow florets and a narrowly cylindrical involucre, and narrowly ellipsoid achenes with 5–7 ribs on either side (Shih and Kilian 2011). L. viminea subsp. viminea, L. viminea subsp. chondrilliflora and L. viminea subsp. ramosissima (2n = 18) share many morphological characters although they differ from each other in certain characteristics. For example, L. viminea subsp. chondrilliflora has a beak length as long as ¼–½ of the achene body while L. viminea subsp. viminea and L. viminea subsp. ramosissima have a beak length equal to the achene body. Furthermore, L. viminea subsp. viminea branches only in the upper part of the stem whereas L. viminea subsp. ramosissima branches mostly in the basal part (Feráková and Májovský 1977). According to Koopman et al. (1998), L. viminea from the section Phaenixopus belongs to the tertiary lettuce gene pool, which also contains L. quercina from section Lactucopsis, L. sibirica and L. tatarica from section Mulgedium. In our phylogentic inferences, L. quercina was not included and L. sibirica and L. tatarica form a seperate Clade 4. Wang et al. (2013) using their nrITS1 sequences indicated a tertiary gene pool similar to Koopman’s but showed that L. sibirica and L. tatarica form a well-supported seperate clade using their plastid gene sequences. Hybridization experiments showed that L. viminea is partly fertile with L. virosa (Groenwold 1983) and L. tatarica could be somatically hybridized with L. sativa (Chupeau et al. 1994; Maisonneuve et al. 1995). As the chance of generating fertile seeds from hybrids of L. tatarica and L. sativa is very low in nature (Chupeau et al. 1994; Maisonneuve et al. 1995), we consider L. orientalis and the three L. viminea subspecies as the tertiary gene pool and keep L. sibirica and L. tatarica beyond the tertiary gene pool.
The lettuce gene pool can provide rich genetic resources for improving lettuce growth, e.g. with respect to resistance to abiotic and biotic stresses. For example, L. serriola from the primary gene pool has been proven to possess interesting alleles for acquiring water and fertilizer in soil, increasing germination and seed longevity (Argyris et al. 2005; Johnson et al. 2000; Schwember and Bradford 2010). L. aculeata from the primary gene pool, L. saligna and L. virosa from the secondary gene pool, L. viminea from the tertiary gene pool, and L. tatarica, L. biennis, L. canadensis, L. homblei, L. indica and L. perennis beyond the lettuce gene pool all showed high resistance to downy mildew (Jeuken et al. 2008; van Treuren et al. 2011). These species may provide rich genetic resources for the crop lettuce. L. orientalis, belonging to the tertiary gene pool, could be a potential resource to improve the growth, development and resistance to diseases of the lettuce crop as well.
Clade 2 (The Pterocypsela Clade) (2n = 18 or ?) This clade comprises species mostly distributed in Asia: L. indica [2n = 18, although Lebeda et al. (2004) indicate it is also in Africa based on floras], L. raddeana (2n = 18) and L. formosana (2n = 18; Hand et al. 2009+; Jeffrey 1966). The only exception is L. ugandensis (2n = ?) from Africa. The first three species belonged to the genus Pterocypsela, which was established by Shih (1988b) with type species Pterocypsela indica (L.) Shih. They have some shared characters, such as involucral bracts in 4–5 rows, capitula with 9–25 florets, broadly winged achenes with 1 or 3(5) prominent ribs on either side of the achene body and double pappus (Shih 1988b, 1997). Shih and Kilian (2011) transferred these three Pterocypsela species to Lactuca. Although L. ugandensis is grouped together with these ex-Pterocypsela species, it is depicted without winged achene (Jeffrey 1966; Jeffrey and Beentje 2000). This L. ugandensis specimen could be mis-identified. Therefore we treat it as Lactuca sp. Clade 2 confirms the nrITS-1 and plastid gene trees of Wang et al. (2013) and is also comparable to section Tuberosae (Lebeda et al. 2007, 2009). In addition, L. indica (Indian lettuce) has been cultivated for its edible leaves (Kadereit et al. 2007). Somatic hybridizations between L. sativa and L. indica have shown that a viable callus can be generated but it cannot produce a viable plant (Mizutani et al. 1989). Moreover, L. indica is resistant to downy mildew (van Treuren et al. 2011). Thus, L. indica could be a useful genetic resource for lettuce breeding.
Clade 3 (2n = 16) This clade is composed of L. dolichophylla, L. dissecta and L. tuberosa (BS = 82, PP = 1). The support value between L. dolichophylla and L. dissecta (BS = 99, PP = 1) is even higher. These three species all have a chromosome number of 16 (Shih and Kilian 2011; Vogt and Aparicio 1999). L. dolichophylla and L. dissecta have some shared characters such as capitula with 6–15(20) blue florets and 3–5 ribs on either side of the achene while L. tuberosa has tuberous roots and broadly winged achenes (Hand et al. 2009+; Shih and Kilian 2011). L. dolichophylla and L. dissecta are distributed in Asia, mainly in South Asia and East Asia, whereas L. tuberosa occurs in Asia and Europe (Geltman 2003; Hand et al. 2009+).
Clade 4 (2n = 34, 16) This clade includes L. canadensis (2n = 34) originating from North America, L. tenerrima (2n = 16) and L. inermis (2n = 16). L. inermis 1 (collected in Ghana) is the sister group to L. canadensis, L. tenerrima and L. inermis 2 (collected in Togo) while L. tenerrima and L. inermis 2 is close to each other (BS = 96, PP = 1; Fig. 1). This could be the result of mis-identification of any of the L. inermis accessions or not enough evidence to distinguish these species. The American Lactuca group includes 12 species, 7 of them are endemic with 34 chromosomes (2n = 34) and different relative DNA content (Babcock et al. 1937; Doležalová et al. 2002; Lebeda and Astley 1999). L. tenerrima and L. inermis (treated as L. capensis before) have been shown to cluster together due to their low DNA content while L. canadensis is far away from them as a result of high DNA content (Doležalová et al. 2003). The crosses between L. canadensis and L. tatarica (2n = 18), and between L. canadensis and L. raddeana (2n = 18) can generate self-sterile hybrid plants (Thompson et al. 1941). Other North American Lactuca species, L. graminifolia Michx. (2n = 34), L. floridana (L.) Gaertn. (2n = 34) and L. spicata Hichc. (2n = 34) could be crossed with L. indica, L. laciniata Roth (now treated as L. indica), L. raddeana, and L. tatarica and produce self-sterile or partly fertile hybrid plants (Thompson et al. 1941; Wang et al. 2013). In addition, L. canadensis, L. raddeana and L. indica share a distinctive character, broadly winged achene, from other Lactuca species although their beak length are clearly different. The North American Lactuca species are supposed to have an amphidiploid origin and arose by subsequent crossings, doubling of chromosomes and hybrid stabilization. Their chromosome complement can be represented by the formula AABB (A = 8, B = 9; Feráková and Májovský 1977). Our phylogenetic inferences and all these experimental hybridizations support the assumption that the North American Lactuca species could have a possible origin from the hybridization between Lactuca species with a haploid chromosome number of 8 (e.g. L. tenerrima) and 9 (e.g. L. tatarica, L. raddeana and L. indica).
Clade 5 (2n = 18) This clade comprises L. undulata from the section Micranthae and L. perennis from the section Lactuca subsect. Cyanicae (Lebeda et al. 2007, 2009). L. undulata shares characters with L. perennis, for example, 1–3 ribs per side of achene and beak as long as achene body (Feráková and Májovský 1977; Shih 1997). This close relationship between L. undulata and L. perennis is supported by Wang et al. (2013). According to Lebeda et al. (2007), species in the section Micranthae have a chromosome number of 16, which is not the case for L. undulata. Therefore, we suggest placing L. undulata into the section Lactuca subsect. Cyanicae.
Clade 6 (2n = 18) This clade contains L. tatarica and L. sibirica from Asia. These species are considered to belong to the section Mulgedium (Lebeda et al. 2007, 2009). Shih (1988b) revised the concept of genus Mulgedium (including L. tatarica) and considered Lagedium Soják (only including L. sibirica) as a monospecific genus, based on the absence of a true beaked achene and a weakly compressed achene body. But Shih’s concept of Mulgedium and Lagedium is not accepted by most taxonomists. Shih and Kilian (2011) revised these two genera and transferred these species into Lactuca. L. sibirica is fully fertile with L. tatarica, indicating a close relationship between these two species (Koopman et al. 2001). However, another European L. tatarica 1 is the sister group to Clade 2 (Fig. 1). This accession is the sister group to Clade 2 in the ndhF tree (Figure S5) and the sister group to the whole Lactuca clade in the trnL-F tree (Figure S6). L. indica in Clade 2 can be crossed with L. tatarica, although producing self-sterile seeds (van Treuren et al. 2011). The conflicting positions of L. tatarica accessions could be the consequence of hybridization. More samples and evidence are needed to solve the problem.
Conclusions
This work presents the first molecular phylogeny of Lactuca with representatives of African species and includes the most extensive sampling of Lactuca species analyzed to date. Based on the results of the phylogenetic trees, we draw the following conclusions:
-
1.
The genus Lactuca contains two well-distinguished clades: the crop clade and the Pterocypsela clade. Other North American, Asian and widespread species either form small clades or are mixed with the Melanoseris species. However, we still think Melanoseris and Lactuca are two separate but closely related genera based on previous studies. The newly identified African endemic species could be treated as a new genus, though more evidence is still needed.
-
2.
We confirm the primary and secondary lettuce gene pool and modify the tertiary gene pool concept: adding L. orientalis and three L. viminea subspecies to the tertiary gene pool while excluding L. sibirica and L. tatarica.
-
3.
L. indica, L. orientalis and L. viminea could be useful genetic resources for lettuce breeding.
-
4.
L. undulata should be transferred from section Micranthae to the section Lactuca subsect. Cyanicae based on our molecular data and its chromosome number.
-
5.
There are at least two independent origins of the scandent habit in Lactucinae.
Although the sampling used in this study only covers 34 % of the total known Lactuca species, we provide the most extensive molecular sampling for Lactuca species to date. Until now, most species in Lactuca have never been revised or sequenced since they were published. In the future, we will sample more species and use whole chloroplast genome data to resolve the polytomy in Lactuca.
References
Anonymous (1925) Contributions from the Gray Herbarium of Harvard University. The Gray Herbarium of Harvard University, Cambridge, Mass, USA
Argyris J, Truco MJ, Ochoa O, Knapp SJ, Still DW, Lenssen GM, Schut JW, Michelmore RW, Bradford KJ (2005) Quantitative trait loci associated with seed and seedling traits in Lactuca. Theor Appl Genet 111:1365–1376
Babcock EB, Stebhins GL, Jenkins JA (1937) Chromosomes and phylogeny in some genera of the Crepidinae. Cytologia, Tokyo, Fujii Jub, pp 188–210
Bakker FT, Lei D, Yu J, Mohammadin S, Wei Z, van de Kerke S, Gravendeel B, Nieuwenhuis M, Staats M, Alquezar-Planas DE, Holmer R (2015) Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an Iterative Organelle Genome Assembly pipeline. Biol J Linn Soc. doi:10.1111/bij.12642
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477
Bentham G (1873) Compositae. In: Bentham G, Hooker JD (eds) Genera plantarum. Reeve & Company, London, pp 163–533
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120
Borsch T, Hilu KW, Quandt D, Wilde V, Neinhuis C, Barthlott W (2003) Noncoding plastid trnT-trnF sequences reveal a well resolved phylogeny of basal angiosperms. J Evol Biol 16:558–576
Bremer B, Bremer K, Heidari N, Erixon P, Olmstead RG, Anderberg AA, Källersjö M, Barkhordarian E (2002) Phylogenetics of asterids based on 3 coding and 3 non-coding chloroplast DNA markers and the utility of non-coding DNA at higher taxonomic levels. Mol Phylogenet Evol 24:274–301
Chupeau M-C, Maisonneuve B, Bellec Y, Chupeau Y (1994) A Lactuca universal hybridizer, and its use in creation of fertile interspecific somatic hybrids. Mol Gen Genet 245:139–145
Clark S, Egan R, Frazier P, Wang Z (2013) ALE: a generic assembly likelihood evaluation framework for assessing the accuracy of genome and metagenome assemblies. Bioinformatics 29:435–443
de Vries IM (1997) Origin and domestication of Lactuca sativa L. Genet Resour Crop Evol 44:165–174
Doležalová I, Lebeda A, Janeček J, Číhalíková J, Křístková E, Vránová O (2002) Variation in chromosome numbers and nuclear DNA contents in genetic resources of Lactuca L. species (Asteraceae). Genet Resour Crop Evol 49:383–395
Doležalová I, Lebeda A, Dziechciarková M, Křístková E, Astley D, van De Wiel CCM (2003) Relationships among morphological characters, isozymes polymorphism and DNA variability—the impact on Lactuca germplasm taxonomy. Czech J Genet Plant Breed 39:59–67
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Edgeworth M (1846) Descriptions of some unpublished species of plants from North-Western India. Trans Linn Soc Lond 20:23–91
Feráková V, Májovský J (1977) The genus Lactuca L. in Europe. Univerzita Komenského, Bratislava
Flann C, Greuter W, Hind DJN (2010) Cassini’s Compositae genera: a nomenclatural and taxonomic assessment. Taxon 59:1206–1244
Geltman DV (2003) Magnoliophyta (= Angiospermae) Magnoliopsida (= Dicotyledones). In: Tzvelev NN (ed) Flora of Russia the European part and bordering regions. A.A. Balkema, a member of Swts and Zeitlinger, pp 195–196
Groenwold R (1983) Onderzoek van de relatie tussen Lactuca en Bremia Lactucae. Verslag van een voorlichtingsbijeenkomst voor slaveredelaars (vervolg). Zaadbelangen 37:132
Hahn C, Bachmann L, Chevreux B (2013) Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res 41:e129
Hand R, Kilian N, Raab-Straube EV (2009+) International cichorieae network: Cichorieae portal. http://cichorieae.e-taxonomy.net/portal/node/8. Accessed 17 Feb 2015
Harlan JR (1986) Lettuce and the sycomore: sex and romance in ancient Egypt. Econ Bot 40:4–15
Hartman Y, Hooftman DAP, Schranz ME, van Tienderen PH (2012) QTL analysis reveals the genetic architecture of domestication traits in Crisphead lettuce. Genet Resour Crop Evol 60:1487–1500
Hartman Y, Hooftman DA, Uwimana B, Schranz ME, van de Wiel CC, Smulders MJ, Visser RG, Michelmore RW, van Tienderen PH (2014) Abiotic stress QTL in lettuce crop-wild hybrids: comparing greenhouse and field experiments. Ecol Evol 4:2395–2409
Hoffmann O (1890–1894) Compositae. In: Hoffmann O (ed) Die natürlichen Pflanzenfamilien. Engelmann, Leipzig, pp 87–387
Jeffrey C (1966) Notes on Compositae: i. Kew Bull 18:427–486
Jeffrey C, Beentje HJ (2000) Cichorieae. In: Beentje HJS (ed) Flora of tropical East Africa, Compositae 1. Balkema, Rotterdam, pp 63–108
Jeuken M, van Wijk R, Peleman J, Lindhout P (2001) An integrated interspecific AFLP map of lettuce (Lactuca) based on two L. sativa × L. saligna F2 populations. Theor Appl Genet 103:638–647
Jeuken MJ, Pelgrom K, Stam P, Lindhout P (2008) Efficient QTL detection for nonhost resistance in wild lettuce: backcross inbred lines versus F2 population. Theor Appl Genet 116:845–857
Johnson WC, Jackson LE, Ochoa O, van Wijk R, Peleman J, St. Clair DA, Michelmore RW (2000) Lettuce, a shallow-rooted crop, and Lactuca serriola, its wild progenitor, differ at QTL determining root architecture and deep soil water exploitation. Theor Appl Genet 101:1066–1073
Judd WS, Campbell C, Kellogg EA, Stevens PF, Donoghue MJ (2007) Plant systematics: a phylogenetic approach. Sinauer Assoc, Sunderland
Kadereit J, et al (2007) Asterales: introduction and conspectus. In: Flowering Plants · Eudicots, vol 8, Springer, Berlin. pp 1–6
Karis PO, Eldenäs P, Källersjö M (2001) New evidence for the systematic position of Gundelia L. with notes on delimitation of Arctoteae (Asteraceae). Taxon 50:105–114
Katoh K, Misawa K, Kuma K-I, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Kerbiriou PJ, Stomph TJ, Putten PEL, Van Bueren ETL, Struik PC (2013) Shoot growth, root growth and resource capture under limiting water and N supply for two cultivars of lettuce (Lactuca sativa L.). Plant Soil 371:281–297
Kesseli R, Ochoa O, Michelmore R (1991) Variation at RFLP loci in Lactuca spp. and origin of cultivated lettuce (L. sativa). Genome 34:430–436
Kilian N, Gemeinholzer B (2007) Studies in the Compositae of the Arabian Peninsula and Socotra: 7. Erythroseris, a new genus and the previously unknown sister group of Cichorium (Cichorieae Subtribe Cichoriinae). Willdenowia 37:283–296
Kilian N, Gemeinholzer B, Lack HW (2009) Tribe Cichorieae. In: Funk VASA, Stuessy T, Bayer R (eds) Systematics, evolution and biogeography of the Compositae. IAPT, Vienna, pp 343–383
Kim K-J, Jansen RK (1995) ndhF sequence evolution and the major clades in the sunflower family. Proc Natl Acad Sci USA 92:10379–10383
Koopman WJM, de Jong JH, de Vries IM (1993) Chromosome banding patterns in Lettuce species (Lactuca sect. Lactuca, Compositae). Plant Syst Evol 185:249–257
Koopman WJM, Guetta E, van De Wiel CCM, Vosman B, van den Berg RG (1998) Phylogenetic relationships among Lactuca (Asteraceae) species and related genera based on ITS-1 DNA sequences. Am J Bot 85:1517–1530
Koopman WJM, Zevenbergen MJ, van den Berg RG (2001) Species relationships in Lactuca s.l. (Lactuceae, Asteraceae) inferred from AFLP fingerprints. Am J Bot 10:1881–1887
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Lebeda A, Astley D (1999) World genetic resources of Lactuca spp.: their taxonomy and biodiversity. In: Lebeda A, Kristkova E (eds) Eucarpia leafy vegetables ‘99. Palacky Univ., Olomouc, Czech Republic, pp 81–94
Lebeda A, Doležalová I, Feráková V, Astley D (2004) Geographical distribution of wild Lactuca species (Asteraceae, Lactuceae). Bot Rev 70:328–356
Lebeda A, Ryder E, Grube R, Doležalová I, Křístková E (2007) Lettuce (Asteraceae; Lactuca spp.). In: Singh R (ed) Genetic resources, chromosome engineering, and crop improvement series, vol 3—vegetable crops. CRC Press, Boca Raton, pp 377–472
Lebeda A, Doležalová I, Křístková E, Kitner M, Petrželová I, Mieslerová B, Novotná A (2009) Wild Lactuca germplasm for lettuce breeding: current status, gaps and challenges. Euphytica 170:15–34
Lindqvist K (1960) On the origin of cultivated lettuce. Hereditas 46:319–350
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Yu C, Wang B, Lu Y, Han C, Cheung DW, Yiu S-M, Peng S, Zhu X, Liu G, Liao X, Li Y, Yang H, Wang J, Lam T-W, Wang J (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1:18
Maddison WP, Maddison DR (2015) Mesquite: a modular system for evolutionary analysis. Version 3.02 http://mesquiteproject.org
Maisonneuve B, Chupeau MC, Bellec Y, Chupeau Y (1995) Sexual and somatic hybridization in the genus Lactuca. Euphytica 85(85):281–285
Matoba H, Mizutani T, Nagano K, Hoshi Y, Uchiyama H (2007) Chromosomal study of lettuce and its allied species (Lactuca spp., Asteraceae) by means of karyotype analysis and fluorescence in situ hybridization. Hereditas 144:235–247
Mikel MA (2007) Genealogy of contemporary North American lettuce. HortScience 42:489–493
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, pp 1–8
Missouri Botanical Garden (2014) Index to Plant Chromosome Numbers (IPCN). http://www.tropicos.org/Project/IPCN. Accessed 07 Oct 2014
Mizutani T, Liu XJ, Tashiro Y, Miyazaki S, Shimasaki K (1989) Plant regeneration and cell fusion of protoplasts from lettuce cultivars and related wild species in Japan. Bull Fac Agric Saga Univ 67:109–118
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Särkinen T, Staats M, Richardson JE, Cowan RS, Bakker FT (2012) How to open the treasure chest? Optimising DNA extraction from herbarium specimens. PLoS ONE 7:1–7
Schneider JV, Bissiengou P, Amaral Mdo C, Tahir A, Fay MF, Thines M, Sosef MS, Zizka G, Chatrou LW (2014) Phylogenetics, ancestral state reconstruction, and a new infrafamilial classification of the pantropical Ochnaceae (Medusagynaceae, Ochnaceae s.str., Quiinaceae) based on five DNA regions. Mol Phylogenet Evol 78:199–214
Schwember AR, Bradford KJ (2010) Quantitative trait loci associated with longevity of lettuce seeds under conventional and controlled deterioration storage conditions. J Exp Bot 61:4423–4436
Shih C (1987) On the circumscription of the genus Prenanthes L. and Notoseris Shih: a new genus of Compositae from China. Acta Phytotaxon Sin 25:189–203
Shih C (1988a) Revision of Lactuca L. and two new genera of tribe Lactuceae (Compositae) on the mainland of Asia. Acta Phytotaxon Sin 26:418–428
Shih C (1988b) Revision of Lactuca L. and two new genera of tribe Lactuceae (Compositae) on the mainland of Asia. Acta Phytotaxon Sin 26:382–393
Shih C (1991) On circumscription of the genus Cicerbita Wall., and two new genera of Compositae from Sino-Himalayan region. Acta Phytotaxon Sin 29:394–417
Shih C (1997) Compositae (10), Cichorioideae, Lactuceae. Science Press, Beijing
Shih C, Kilian N (2011) Cichorieae [excl.Tragopogon, Taraxacum, Hieracium and Pilosella]. Science Press & St Louis: Missouri Botanical Garden, Beijing, pp 233–238
Simmons MP, Ochoterena H (2000) Gaps as characters in sequence-based phylogenetic analyses. Syst Biol 49:369–381
Staats M, Cuenca A, Richardson JE, Vrielink-van Ginkel R, Petersen G, Seberg O, Bakker FT (2011) DNA damage in plant herbarium tissue. PLoS ONE 6:e28448
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Stebbins GL (1937a) Critical notes on Lactuca and related genera. J Bot 75:12–18
Stebbins GL (1937b) The scandent species of Prenanthes and Lactuca in Africa. Bulletin du Jardin botanique de l’État a Bruxelles 14:333–352
Stebbins GL (1939) Notes on some Indian species of Lactuca. Indian For Bot 1:237–245
Stebbins GL (1940) Studies in the Cichorieae: Dubyaea and Soroseris, endemics of the Sino-Himalayan Region. Mem Torrey Bot Club 19:1–76
Stover BC, Muller KF (2010) TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform 11:7
Swofford D (2003) PAUP*: phylogenetic analysis using parsimony (* and other methods). In: Associates S (ed) 4.0b 10 edn. Sunderland Massachusetts USA
Taberlet P, Coissac E, Pompanon F, Gielly L, Miquel C, Valentini A, Vermat T, Corthier G, Brochmann C, Willerslev E (2007) Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res 35:e14
Thiers B (2011) Index Herbariorum: a global directory of public herbaria and associated staff. New York Botanical Garden’s Virtual Herbarium. http://sweetgum.nybg.org/ih/. Accessed 17 Feb 2015
Thompson RC, Whitaker TW, Kosar WF (1941) Interspecific genetic relationships in Lactuca. J Agric Res 63:91–107
Tuisl G (1968) Der Verwandtschaftskreis der Gattung Lactuca L. im iranischen Hochland und seinen Randgebieten. Ann Nat Hist Mus Wien 72:587–638
Uwimana B, Smulders MJ, Hooftman DA, Hartman Y, van Tienderen PH, Jansen J, McHale LK, Michelmore RW, Visser RG, van de Wiel CC (2012) Crop to wild introgression in lettuce: following the fate of crop genome segments in backcross populations. BMC Plant Biol 12:43
Vaidya G, Lohman DJ, Meier R (2011) SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27:171–180
van Treuren R, van der Arend AJM, Schut JW (2011) Distribution of downy mildew (Bremia lactucae Regel) resistances in a genebank collection of lettuce and its wild relatives. Plant Genet Resour 11:15–25
Vogt R, Aparicio A (1999) Chromosome numbers of plants collected during Iter Mediterraneum IV in Cyprus. Bocconea 11:117–168
Wang ZH, Peng H, Kilian N (2013) Molecular phylogeny of the Lactuca alliance (Cichorieae subtribe Lactucinae, Asteraceae) with focus on their Chinese centre of diversity detects potential events of reticulation and chloroplast capture. PLoS ONE 8:e82692
Wei Z, Julkowska MM, Laloë J-O, Hartman Y, de Boer G-J, Michelmore RW, van Tienderen PH, Testerink C, Schranz ME (2014) A mixed-model QTL analysis for salt tolerance in seedlings of crop-wild hybrids of lettuce. Mol Breed 34:1389–1400
Whitaker TW, Thompson RC (1941) Cytological Studies in Lactuca. Bull Torrey Bot Club 68:388–394
Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20:3252–3255
Yu Y et al (2015) RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Mol Phylogenet Evol 87:46–49
Zhang J-W, Nie Z-L, Sun H (2009a) Cytological study on the genus Syncalathium (Asteraceae-Lactuceae), an endemic taxon to alpine scree of the Sino-Himalayas. J Syst Evol 47:226–230
Zhang NW, Lindhout P, Niks RE, Jeuken MJW (2009b) Genetic dissection of Lactuca saligna nonhost resistance to downy mildew at various lettuce developmental stages. Plant Pathol 58:923–932
Zhang J-W, Nie Z, Wen J, Sun H (2011) Molecular phylogeny and biogeography of three closely related genera, Soroseris, Stebbinsia, and Syncalathium (Asteraceae, Cichorieae), endemic to the Tibetan Plateau, SW China. Taxon 60:15–26
Zohary D (1991) The wild genetic resources of cultivated lettuce (Lactuca sativa L.). Euphytica 53:31–35
Acknowledgments
Thanks to China Scholarship Council (http://en.csc.edu.cn/) for providing funding to Z.W. The contribution from SYNTHESYS Joint Research Activities 4 (JRA4: Plants/fungi herbarium DNA) were supported by European Union-funded integrated Activities grant awarded to Dr. F.T.B. and Dr. W.G. Berendsohn. We appreciate Dr. J.J. Wieringa, Dr. MJW (Marieke) Jeuken and Dr. N.K. for providing plant materials for DNA extraction. Thanks to the anonymous reviewer for identifying herbarium specimen in this study.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
Cite this article
Wei, Z., Zhu, SX., Van den Berg, R.G. et al. Phylogenetic relationships within Lactuca L. (Asteraceae), including African species, based on chloroplast DNA sequence comparisons. Genet Resour Crop Evol 64, 55–71 (2017). https://doi.org/10.1007/s10722-015-0332-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-015-0332-5